
Abstract
Analytical and numerical modeling has emerged as a valuable tool for planning and designing groundwater remediation systems. Models have been used in a variety of settings including (1) research into the fundamental processes controlling chlorinated solvent fate and transport, (2) methods for integrating information on site hydrology, geology, contaminant distribution, transport and fate, and (3) applied aspects of plume management and remediation system design. This chapter focuses on currently available models commonly used by practitioners for simulating dissolved chlorinated solvent plumes and includes a brief summary of modeling principles, mathematical expressions useful for representing biodegradation processes, methods for representing dissolved contaminant release from source areas and case studies of models applied to sites.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
6.1 Introduction
Analytical and numerical modeling has emerged as a valuable tool for planning and designing groundwater remediation systems. Models have been used in a variety of settings including (1) research into the fundamental processes controlling chlorinated solvent fate and transport, (2) methods for integrating information on site hydrology, geology, contaminant distribution, transport and fate, and (3) applied aspects of plume management and remediation system design. This chapter focuses on currently available models commonly used by practitioners for simulating dissolved chlorinated solvent plumes and includes a brief summary of modeling principles, mathematical expressions useful for representing biodegradation processes, methods for representing dissolved contaminant release from source areas and case studies of models applied to sites. Key challenges that face the groundwater remediation industry from a modeling standpoint include:
-
Modeling the complex biological reactions involved in biodegrading chlorinated solvents and being able to simulate the natural attenuation capacity of the groundwater aquifer
-
Understanding the processes controlling remediation of chlorinated solvents and modeling them
-
Predicting remediation impacts using simplified and/or complex models, and
-
Evaluating technological efficiencies and performance of technologies used to remediate dissolved chlorinated solvent plumes.
Researchers have been addressing these challenges and developing analytical and numerical models that simulate the fate and transport of chlorinated solvents in the subsurface. Analytical models provide exact solutions to the fate and transport equations whereas numerical models are used to generate estimated solutions that are within an acceptable accuracy relative to the exact solution. This chapter describes these advances in modeling and the resulting models that have been used to simulate chlorinated compounds and their remediation.
6.2 Fate and Transport Modeling
Chlorinated solvents released to the subsurface undergo a number of physical, chemical and biological changes as they distribute into the aquifer matrix and groundwater flowing through the matrix. Groundwater flow within the matrix causes the dissolved chlorinated solvents to be transported (or advected) along the flow paths. The heterogeneities and nonuniformity of the aquifer matrix cause microscale spreading, commonly referred to as dispersion. Microbial populations that naturally exist in subsurface media can, given the right conditions, metabolize (break down) chlorinated solvents into other organic compounds or effect their complete mineralization. Because of their organic nature, chlorinated solvents can attach (partition or sorb) to the organic fraction of the aquifer matrix, thus slowing their transport in the dissolved phase. In addition, chlorinated solvents can undergo abiotic reactions in the subsurface that affect their fate and transport.
This section will focus on the basics of fate and transport modeling. More detailed and thorough discussions on the physical, chemical, biological and abiotic mechanisms and processes affecting chlorinated solvents can be found in other references including Bedient et al. (1999), Domenico and Schwartz (1998), and Wiedemeier et al. (1999). Information on chlorinated solvents in dense nonaqueous phase liquid (DNAPL) form can be found in Schwille (1988) and Cohen and Mercer (1993). Detailed discussions of modeling principles are available in Zheng and Bennett (1995), Anderson and Woessner (1992) and Bear and Verrujit (1987).
6.2.1 Basic Fate and Transport Equations
In general, the change in dissolved concentrations in an aquifer matrix due to advection and dispersion in mathematical form in one dimension (1-D) is given by:
where C is the contaminant concentration (M/L3), t is time, \(D_x\) is the coefficient of hydrodynamic dispersion (L2/T), \(v_x\) is the average seepage velocity in groundwater (L/T), and \(x\) is distance along the flow path. The symbols M, L and T refer to standard units—M is Mass (generally expressed in milligrams [mg] or micrograms [µg]), L is Length (usually expressed in meters [m] or centimeters [cm]), and T is time (generally expressed in days or seconds). Thus, L2 represents area and L3 represents volume.
Incorporating sorption into the 1-D advection-dispersion equation is accomplished through the use of a retardation factor (assuming linear reversible sorption):
where \(R\) is the coefficient of retardation (dimensionless).
In its simplest form, incorporating biodegradation as a first-order reaction into the 1-D advection-dispersion equations results in:
where \(\lambda\) is the first-order biodegradation rate coefficient (1/T). The two terms, \({{\partial C} \over {\partial t}}\) and \(\lambda C\), do not incorporate a velocity term; thus, they are multiplied by R, the retardation coefficient, to reflect the effects of sorption.
Solving Equations 6.1, 6.2 and 6.3 requires knowledge about the system that is being modeled. This is usually termed initial and boundary conditions. Initial conditions specify the groundwater contaminant concentrations at the beginning of the simulation (or model run). Boundary conditions, on the other hand, specify the value of the dependent variable (dissolved concentration in this case), or the value of the first derivative of the dependent variable (flux of dissolved concentration), along the boundaries of the system that is being modeled. As an example, if an experiment in a laboratory column is being modeled with a continuous constant source input, initial conditions would include:
Equation 6.4 means that the initial concentration in the column at any \(x\) before the experiment starts (i.e., \(t = 0\)) is zero. Boundary conditions, on the other hand, define concentrations at either end of the column and would include:
Equations 6.5 and 6.6 basically indicate that a concentration of C0 enters the column on one end and that the concentration beyond the column is zero at all times. Thus, the conditions associated with the particular scenario at hand are defined.
Once the initial and boundary conditions have been specified, mathematical and numerical techniques are used to solve Equations 6.1, 6.2 or 6.3, subject to initial and boundary conditions, such as those given by Equations 6.4 through 6.6.
As an example, the analytical (or exact answer) solution to Equation 6.3 for an instantaneous source (a slug of chlorinated solvent) released into the groundwater is given by:
where \(M\) is the injected mass per unit cross-sectional area. Figure 6.1 shows the results from a scenario analysis using Equation 6.7.

Concentrations as a function of time and distance from an instantaneous release in groundwater.
The boundary condition shown in Equation 6.5 designates what is known as a “constant concentration” boundary condition. Other types of boundary conditions used to define sources of chlorinated solvents to groundwater include a “constant flux” boundary condition in which the mass flux (M/V/T) is defined. Constant concentration boundary conditions are typically used to represent dissolution from residual sources, while mass flux boundary conditions are used to represent leakage of chemicals into the groundwater due to the combined effects of infiltration, fluctuations of the groundwater table that cause dissolution from residual sources, and continuous spillage events from tanks, ponds, pits, etc. The difficulty in modeling these processes, as will be discussed in Section 6.4, lies in understanding source behavior over time and estimating the mass flux rate that is “feeding” that groundwater plume.
The advection-dispersion equation also can be written in more than one dimension. In three-dimensional (3-D) terms, the equation is expressed as:
where \(D_y\) and \(D_z\) are the coefficients of hydrodynamic dispersion (L2/T) in the y and z dimensions.
It should be noted that Equation 6.8 has a 1-D velocity and that is the velocity in the x-direction (or the direction of groundwater flow), and it is only 3-D in the context of dispersion, i.e., dispersion is in the x-, y-, and z-directions. This simplifies the solution greatly and allows Equation 6.8 to be solved analytically once the initial and boundary conditions have been defined. The solution only requires the dispersion coefficients in the transverse (y-direction), and vertical (z-direction) over and beyond the data requirements of the 1-D solution.
One recent issue has been the accuracy of the Domenico solution (Domenico, 1987), one of the most commonly used solutions for solving the advection-dispersion equation (Equation 6.8) analytically. West et al. (2007) suggested that the Domenico solution not be used because of the error involved in approximation terms employed in the solution. Other researchers (e.g., Falta et al., 2005b; Guyonnet and Neville, 2004; USEPA, 2007) have concluded that the Domenico solution yields reasonably accurate results for most of the cases where it is typically applied.
Unlike Equation 6.8, “fully 2- or 3-D” models require defining velocities in the x, y, and/or z directions along with dispersion in two- or three dimensions. In this instance, the 2-D advection-dispersion equation is given by:
where \(q_s\) is the discharge rate of the source or sink into the aquifer, \(\theta\) is the effective porosity, and \(C_s\) is the concentration associated with the source or sink. As can be seen from Equation 6.9, the input data requirements are much greater in this case and include velocities and dispersion coefficients in two dimensions and source or sink data, as well as reaction and sorption rates. When using a 2-D model, it is assumed that the dissolved chlorinated solvents are completely mixed in the vertical z-dimension or the thickness of the aquifer and that the solvent concentrations measured in monitoring wells are uniform across the aquifer depth. Furthermore, solving Equation 6.9 requires either solving the flow equation in 2-D to obtain velocities or using a groundwater flow model to estimate the velocity field. Solving Equation 6.9 also involves relying on numerical or approximation methods that discretize space and time and have error associated with them (due to their approximating nature). The key advantage of using this modeling approach, however, stems from the ability it provides for simulating the varying conditions across the site, such as subsurface heterogeneities and changes in sources and sinks or reactions over time.
Equations 6.3, 6.8 and 6.9 make use of a simplified biodegradation reaction term, \( - \lambda C\). This type of expression is used when biodegradation is assumed to be a first-order process such that the change in concentration over time due to the reaction is a function of the concentration, or:
The resulting concentration from this type of biodegradation kinetic expression is shown in Figure 6.2 for various values of \(\lambda\). The rate constant, \(\lambda\), is typically expressed in terms of a reaction half life, the time required for the mass of the biodegrading compound to decrease to half the original mass. The solution to Equation 6.10 is an exponential function of the form:

Concentration change as a function of time for various values of \(\lambda\).
where \(C_0\) is the starting concentration of the chemical at time = 0. The half life of the chemical is given by:
The simplified first-order expression given by Equation 6.10 is popular among modelers and practitioners because it requires only one variable to evaluate the effect of biodegradation on a given chlorinated solvent plume, and that is the rate constant, \(\lambda\). Many acknowledge, however, that estimating this variable from field data is quite complex because field concentrations reflect the combined effect of many processes, such as biodegradation, advection, dispersion and sorption, and it is difficult to separate them out without adequate data gathering and specialized testing. Furthermore, estimating \(\lambda\) from laboratory studies is problematic as well since laboratory experimental conditions are simplified and do not reflect the complexity and the conditions encountered in the subsurface.
A U.S. Environmental Protection Agency (USEPA) Remedial Technology Fact Sheet (Newell et al., 2002) presents methods to estimate biodegradation rates and cautions that different attenuation rates calculated by different methods represent different processes at sites. A concentration vs. time rate represents the attenuation of source materials and is not directly related to a biodegradation rate. Biodegradation rates are lumped together with dispersion in concentration vs. distance rate calculations. Specific methods were identified to calculate biodegradation rates, as discussed below.
Buscheck and Alcantar (1995) derived a relationship that allows calculation of approximate biodegradation rate constants using field data. Their method assumes, however, that a dissolved plume has reached steady-state conditions, an assumption that is not always realized or true for chlorinated solvent plumes. The first-order rate constant is calculated as:
where \(\alpha _x\) is the dispersivity (the dispersivity is related to the dispersion coefficient \(D_x\) such that \(D_x = \alpha _x v_x\)), and \({k \over {v_x }}\) is the slope of the line formed by making a log-linear plot of contaminant concentration versus distance downgradient along the flow path. Examples of how to apply this method can be found in Buscheck and Alcantar (1995), Wiedemeier et al. (1996) and Chapelle et al. (1996).
In addition to Equation 6.13, Wiedemeier et al. (1999) developed a method that estimates \(\lambda\) by comparing concentrations for the chlorinated compound to those of a tracer that might be present at the site in the subsurface. This tracer could be an inorganic compound (such as chloride), a recalcitrant compound (potentially tri-methyl benzenes) or calibration of groundwater solute transport models to field data. More recently, methods involving carbon and chlorine isotope analysis have been used to quantify biodegradation processes (e.g., Hirschorn et al., 2007; Philp et al., 2007).
Additionally, a number of researchers have catalogued, measured or reported biodegradation first-order rates from the literature for chlorinated compounds. Table 6.1 presents data compiled by Suarez and Rifai (1999) from both laboratory and field studies. Table 6.2 shows decay rates calculated from field sites as determined from calibrations of the BIOCHLOR model (Aziz et al., 2000b).
Another popular biodegradation expression used in modeling because of its simplicity is the zero-order expression. In this expression, the change in concentration as a function of time due to biodegradation is a function of the reaction rate constant:
where \(\kappa\) is the zero-order rate constant (1/T). Suarez and Rifai (1999) provided data on zero-order rates for chlorinated solvents from the general literature.
More sophisticated kinetic approaches for modeling the biodegradation of chlorinated solvents involve the use of Monod kinetics, and incorporate the limitations of multiple substrates, and inhibitory processes. These approaches are discussed in more detail in Section 6.3.
Irrespective of the kinetic expression used to model biodegradation of chlorinated compounds, it is critical to recognize that source behavior over time has a significant influence on the observed concentrations within the plume. A representative site model needs to incorporate adequate expressions to model both processes: biodegradation and source behavior. Typically, residual sources in chlorinated plumes have been described using a starting concentration and a source “decay” rate much like the first-order reaction rate constant discussed earlier and shown in Equation 6.10.
Hausman and Rifai (2005) elucidated the effects of the biodegradation rate constant and the source decay constant on perchloroethene (PCE) concentrations in groundwater, as well as their combined effects. Their analysis considered a relatively short timeframe of 10 years and a longer duration of 100 years. Figure 6.3a illustrates the change in PCE concentrations due to biodegradation, and Figure 6.3b presents the concentration changes due to source decay for the same scenario. Figure 6.4 shows the resulting PCE groundwater concentrations

Model simulation of PCE downgradient concentrations as a function of (a) biodegradation and (b) source decay (Hausman and Rifai, 2005).

Model simulation of PCE downgradient concentrations as a function of biodegradation and a constant source decay value of 0.03 1/yr (Hausman and Rifai, 2005).
for a biodegrading plume with source decay. It can be seen from Figure 6.3a that biodegradation for both short and long durations has a significant impact on concentrations downgradient from the source area, but has no impact on source concentrations (when assuming a constant non-decaying source). Figure 6.3b, on the other hand, illustrates that source decay affects the concentrations in the source area but has little short-term or long-term impact on downgradient concentrations. The combined effect of these two processes (source decay and biodegradation), however, cause a plume to be shorter and have lower concentrations downgradient from the source. Thus, in modeling chlorinated solvents, it is important to adequately define these processes.
6.2.2 The Modeling Process
Modeling can be a useful analytical tool to better understand contaminant behavior and responses to remediation technologies. However, it is important to recognize that applying a model to a field problem involves uncertainty due to subsurface heterogeneities and the spatial and temporal variations of the physical, chemical and biological processes involved. In addition, mistakes in model applications are common, and models may be misused or applied inappropriately. However, the benefits of learning and applying models outweigh the drawbacks because models provide:
-
An ability to simulate a system or its components
-
An understanding of how a process or mechanism affects contaminant distributions in groundwater
-
A design tool that can be used to evaluate different remediation designs, and
-
An “evergreen” process that can be used to continuously update our knowledge about a site.
The modeling process and models have been described as “garbage in–garbage out.” This is because when model set-up, calibration and validation are not rigorously undertaken, model results are suspect and of little value to the stakeholders. Thus, the key controlling factor in undertaking “successful” model development is careful adherence to the modeling process including (1) developing a conceptual model, (2) articulating the modeling objective, (3) analyzing site data to estimate model variables, (4) calibrating and validating the model and, finally, (5) using the model for predictive analysis. The modeling process also involves assessing the uncertainty or confidence in the model results. Uncertainty assessments can be accomplished in a number of ways, as will be seen later, but it is common for modelers to use sensitivity analysis to identify the key model variables that have the most influence on model results. A sensitivity analysis is typically undertaken for the calibrated model as well as the predictive model. The following sections further describe the modeling process.
6.2.2.1 Conceptual Model
The key step to modeling fate and transport at a field site is to gather and analyze all of the relevant information for the site. Then a conceptual model is developed that integrates the collected information and describes many of the variables and processes involved at the site. This process includes the introduction of simplifying assumptions and interpretations of the flow and transport regimes. Preliminary calculations and simple analytical models can be used in this step to help develop a conceptual understanding. For example, Darcy velocities can be estimated and used to develop estimates of plume length if the starting date of contamination is known. The observed plume length then can be compared to these estimates and interpretations regarding chemical and biological processes can be made. If, for example, the observed plume is shorter than expected, it may be due to the combined effects of sorption and biological or abiotic reactions. These processes would then need to be included in the model that is being developed.
Conceptual model development includes defining the hydrogeologic environment (e.g., homogeneous, heterogeneous, layered), the flow regime (confined, unconfined, direction and rate of flow), the source of the contamination (leak or spill start date, what was spilled and how much), and the chemical and biological properties of the chemicals involved (sorption and biodegradation characteristics, for example). Conceptual model development also includes interpreting the groundwater level data, groundwater chemical data and subsurface matrix data and any other types of information gathered at the site. Monitoring well contaminant concentrations and plume maps can be used to develop estimates of plume length over time and attenuation rates within the plume.
The importance of developing an appropriate conceptual model for a site cannot be overstated. The conceptual model is used to guide model selection, model development, parameter estimation, calibration and validation, sensitivity analyses and prediction. Without an appropriate conceptual model, the exercise of modeling is lacking in context and validity. It should be noted that the conceptual model is not static; it is expected to evolve and change as more data are gathered and knowledge of a given site increases. However, Bredehoeft (1994) discusses the “surprise” factor, where many (i.e., perhaps 20–30%) groundwater conceptual models are rendered invalid by new data. Overall, Bredehoeft concludes that developing conceptual models of groundwater systems involves uncertainty “… that often is not widely recognized” and that this uncertainty is magnified in long-term predictions using groundwater models. Regardless, modeling without an appropriate conceptual model leads to even greater uncertainty and less confidence in modeling results.
6.2.2.2 Goals of Model Development
The first question that needs to be answered is: What is the purpose of the modeling? It is important to ascertain why the modeling is being done. Typical objectives include (1) understanding a phenomenon or process and how it might affect contaminant fate and transport, (2) establishing likely scenarios that caused the observed contaminant concentrations at a site (source strength, duration of spill, etc.) and (3) predicting future contaminant distributions either under existing conditions or with engineered remediation systems. Defining the objective of the modeling has a strong bearing on model selection, discretization of the modeled domain and the level of effort that must be expended in the modeling study. Additionally, stating the goal for model development defines the expectations and allows model results to be used most appropriately.
6.2.2.3 Data Analysis and Parameter Estimation
The success of model development and application in a given situation relies largely on developing appropriate estimates for the model variables. Information gathered from a site or an experiment should be used to develop these estimates. For example, water level data from monitoring wells or piezometers can be analyzed to estimate the groundwater flow direction and rate. Variables such as hydraulic conductivity, aquifer thickness and flow boundary conditions must then be estimated to calibrate the flow model such that the model-generated flow field matches the observed one.
Once data from a site have been analyzed and integrated into a conceptual model, as discussed earlier, it becomes necessary to begin building the transport model. This requires selecting the dimension of the analysis (1-, 2- or 3-D). It also requires discretizing the spatial and temporal domains. Spatial discretization involves selecting a model grid to represent the site while temporal discretization involves defining what timeframe will be modeled and at what resolution. These are very important considerations that have to be determined on a site-specific basis and depend on which model is being used. Suffice it to say that the spatial domain must be large enough to account for the sources and sinks of water and chemicals in the aquifer that affect the observed plume. The temporal domain depends again on the goal of the modeling and the conceptual model for the site and whether invasive remediation or natural attenuation is to be simulated.
In addition to the spatial and temporal considerations discussed above, initial and boundary conditions must be defined. These conditions also vary from site to site and include considerations such as sources and sinks of water and chemicals into the selected grid, and contaminant conditions within the aquifer prior to the modeled time period. Appropriate selection of initial and boundary conditions eliminates a common problem often encountered in modeling- use of a site model that does not adequately represent the site-specific conditions.
6.2.2.4 Calibration
After the model has been set up and initial estimates of the input parameters have been developed, the model must be calibrated against the site data. In most (if not all) instances, the parameter estimates must be altered until the model’s simulated values satisfactorily match their measured counterparts. The calibration process is one of the most critical and valuable steps of modeling. Calibration may be formally accomplished through the use of optimization techniques or less formally by trial and error. Both approaches have advantages and disadvantages, but it is important to understand that the calibration process is not unique. In other words, there are likely many sets of parameter estimates that would produce an acceptable calibration. In one classic study, Freyberg (1988) assigned nine groups of graduate students the same groundwater modeling assignment. Group predictions varied significantly, however, leading Freyberg to conclude that “good calibration did not lead to good prediction.” In a more recent study, Saiers et al. (2004) presented a case study where three groundwater models were constructed using different types of groundwater data. The three models gave similar predicted results for groundwater level, but not for flow. Finally, Konikow and Bredehoeft (1992) concluded that “groundwater models cannot be validated, but only tested and invalidated.”
The goal in calibration is to select the set of values that most closely represents the likely values of the parameters for the site in question. This, of course, requires identifying the measure of success that will be used to determine if the model is adequately calibrated or not. As an example, in calibrating the model to simulate the observed flow field at a site, a number of goodness-of-fit measures can be used: (1) the modeled flow field can be visually or statistically compared to the contoured flow field from water level data, (2) the modeled water levels at the monitoring wells can be compared to the observed water levels or (3) a combination of the two can be used. When comparing modeled water levels to measured ones or modeled water contours to their observed counterparts, the modeler will need to select a method for estimating the error and an acceptable target for the error (<5% error, etc.). In this way, whether by trial and error or automated calibration, the “best” set of parameter estimates can be selected and the model calibrated.
Recently, there has been a trend to use automatic calibration techniques. Carrera et al. (2005) discussed the overall calibration approach and methods to improve the calibration process. Doherty (2003) proposed a calibration approach for groundwater models using a system of “pilot points” that make nonlinear parameter estimation programs, such as PEST (Doherty, 2005), more flexible and easier to implement. Hunt et al. (2007) compared simple groundwater modeling approaches to more complicated models and concluded that the regularized inversion, which includes many more parameters than conventional approaches, has significant advantages for the modeling process. Kelson et al. (2002) presented the opposite conclusion, where a modest model based on less than 10 parameters yielded similar results to much more complex codes.
6.2.2.5 Validation
The process of validation is just as important as calibration because it allows for greater confidence in model results. Basically, validation involves using a second set of data that is distinct from the data set used for calibration and that was collected at a later point in time than the calibration data set. The calibrated model is then run for the time period between the calibration and validation data sets in order to predict contaminant concentrations for the validation timeframe. Model results are then compared to the validation data and an assessment of closeness of fit is made. If the results are acceptable (i.e., within a predefined margin of error), the validated model can be used for prediction analysis. However, if the validation results are unacceptable, then a recalibration of the model is required using the two datasets (calibration and validation) in order to fine-tune the parameter estimates.
In some instances, validation data sets are not available. In this case, modelers have to rely on a sensitivity analysis to determine which variables have the most impact on the model results and to define and quantify the level of confidence in the model output.
6.2.2.6 Prediction
Once a model has been calibrated and validated, it can be used to predict future contaminant distributions under natural or engineered remediation conditions. Predictions under natural conditions typically involve running the model beyond the present time into the future to determine plume dimensions and concentrations over time. This type of simulation can also include scenarios such as reducing source concentration or mass due to source control technologies. In the same way that a sensitivity analysis is undertaken for the calibrated model, it is necessary to complete a sensitivity analysis for the predictive model to determine the confidence in model results.
Predicting future concentrations using remediation technologies can involve simulating injection and pumping or a change in the flow conditions, as well as possibly a change in source concentrations or flow rates. Caution must be exercised in this instance, because of the uncertainties involved in simulating engineered remediation systems and their performance. For example, if the engineered remediation includes pumping water from the aquifer, then a secondary process of calibration may be required to ascertain that the model adequately represents pumping conditions (e.g., the model can be used to match data from a pumping test in the aquifer). Additionally, it is important to keep in mind that pumping and injection of water into the plume may have an effect on the source definition used in the model, which needs to be recognized. Thus, it is just as important to assess or quantify the confidence in the model results in the predictive mode as it is in the calibration mode.
6.3 Modeling Biodegradation, Sorption and Abiotic Reactions of Chlorinated Solvents
Chlorinated solvents can be mineralized and/or partially biodegraded by microorganisms under aerobic and anaerobic conditions, as demonstrated in numerous studies (e.g., Hopkins and McCarty, 1995; Wilson and Wilson, 1985; Fogel et al., 1986; Little et al., 1988; Malachowsky et al., 1994; Wackett et al., 1989; Vanderberg et al., 1995; Nelson et al., 1988; Bouwer and McCarty, 1983; and Folsom et al., 1990). It is generally accepted that enzymes involved in metabolizing a primary substrate such as methane or propane under aerobic conditions or natural organic compounds under anaerobic conditions catalyze the biodegradation of chlorinated solvents (Zhang et al., 1996). However, the biodegradation process may be limited or constrained by a number of factors, including biological limitations and environmental conditions in the subsurface. Microbial considerations include activity loss of critical enzymes, energetic needs for chlorinated biodegradation, metabolic patterns in mixed cultures, and accumulation and disappearance of metabolic intermediates. Subsurface considerations include the carbon content, pH, redox, the dissolved contaminant concentrations, and the presence of several chlorinated solvents with differing biodegradation characteristics, among others.
Key degradation processes for chlorinated solvents include the following (Truex et al., 2006; Wiedemeier et al., 1999):
-
Aerobic cometabolism
-
Anaerobic cometabolism
-
Aerobic direct metabolism
-
Anaerobic direct metabolism
-
Dehydrochlorination (abiotic)
-
Abiotic hydrolysis
-
Dichloroelimination (biotic)
-
Reductive dechlorination (hydrogenolysis)
Truex et al. (2006) used this list of reactions to show detailed degradation charts for the families of chlorinated ethanes, chlorinated ethenes and chlorinated methanes for aerobic, anoxic and anaerobic geochemical environments.
While mathematical models have been developed to simulate the biodegradation of chlorinated solvents in groundwater, including some of the considerations discussed above, it is important to keep in mind that the models developed to date are far from being able to simulate the complexity of the biodegradation process under field conditions. It is also important to note that appropriately describing the source term for a dissolved chlorinated plume is just as important, if not more important, than fully describing the complexity of the biodegradation process. Additionally, a critical consideration in modeling chlorinated solvents is the need to balance the complexity of the biodegradation model with the available data to support model development and application.
In addition to the first-order model presented in Equation 6.10, Monod kinetics has been a typical starting point for many of the mathematical models developed to date. Monod kinetics relates the transformation rate of the chlorinated solvent to its concentration and the concentration of the microorganisms (whereas the first-order model relates the concentration change to the concentration of the chemical):
where \(C\) is the substrate concentration, \(t\) is time, \(X\) is the microbial concentration, \(k\) is the maximum specific utilization rate of the substrate, and \(K\) is the half-saturation constant for the substrate.
Equation 6.15 is based on the assumption that the reaction is limited by the one substrate; however, in practice, there are multiple substrates involved in the biodegradation of chlorinated solvents, necessitating the use of multiple-substrate Monod kinetic expressions:
where \(C_e\) is the concentration of a limiting electron acceptor or nutrient, and \(K_e\) is the half-saturation constant for the electron acceptor or nutrient. Widdowson et al. (1988) and Borden and Bedient (1986) used this approach to develop expressions for aerobic and anaerobic biodegradation of organic compounds.
When competitive inhibition between two substrates is occurring (for example, the chlorinated solvent and a primary substrate), the Monod kinetic expressions are modified to:
where \(C_1\) and \(C_2\) represent the primary substrate and the chlorinated solvent.
The Monod kinetic expression also has been modified to simulate cometabolic degradation of the chlorinated solvents. The reader is referred to Alvarez-Cohen and Speitel (2001), Ely et al. (1997) and Zhang and Bajpai (2000) for detailed discussions on the aerobic and anaerobic kinetic expressions used to simulate cometabolism.
Several issues are noted from Equations 6.16 through 6.18. First, the expression in Equation 6.16 typically replaces the \( - \lambda C\) expression in the fate and transport equation for the chlorinated compound (for example, Equation 6.3). This means, however, that one or more fate and transport equations (in addition to that for the chlorinated compound) will need to be solved to determine the concentrations in groundwater of the limiting substrate(s). Second, Equation 6.18 requires defining the microbial concentration, so unless the modeler assumes a constant microbial population concentration, it will be necessary to simulate the growth, death and transport of the microbial population. Thus, it can be seen that using Monod kinetics adds a layer of complexity and increases the data requirements.
A third and equally important consideration is the different time scales between transport and biodegradation. In groundwater, contaminant transport times can range from a few years to several decades. On the other hand, biodegradation half-lives can range from days or weeks to years or even decades. Hence, selection of an appropriate modeling time step is important to increase the probability that modeling results will be accurate. When the reaction half life is much smaller than the transport time, the time step used in modeling should be at the same scale as the reaction half life to yield accurate results. Borden and Bedient (1986) and Rifai et al. (1988) addressed this issue for petroleum hydrocarbons by assuming that the biodegradation reaction is almost instantaneous, thus eliminating the need for very small time steps that increase runtimes. Their method, however, introduces error when used for slow reactions because it assumes that substrates are used in an instantaneous manner, which would not be the case when the reaction is, in fact, slow and occurs at an equivalent time scale as transport.
New models also have been developed to simulate new ideas about sorption and desorption processes. Many numerical models allow the user to select between a linear, Freundlich or Langmuir isotherm (see Chapter 4), while analytical models are often limited to the linear expression. One exception is the RT3D numerical solute transport model, which has the capability to simulate rate-limited sorption processes, where desorption is controlled by a mass-transfer limitation step (Clement, 1997). Similarly, the model developed by Chen et al. (2004) includes a dual-equilibrium desorption approach (Chen et al., 2002) to model the availability effects on desorption processes.
Finally, there has been new research on the potential for abiotically mediated reductive dechlorination of chlorinated solvents by iron in the ferric (Fe(II)) state (Ferrey et al., 2004). This abiotic dechlorination occurs under anoxic conditions when Fe(II)-bearing minerals (magnetite, pyrite, green rust, mackinawite) release electrons for reduction of contaminants via a surface-mediated reaction. Because Fe(II) is relatively soluble under typical groundwater conditions (neutral pH), measurements of total Fe(II) in a soil matrix can include both dissolved and mineral forms. However, oxidation of dissolved Fe(II) is not thought to be capable of reductive dechlorination of contaminants of concern (COCs) in the absence of a solid phase (Lee and Batchelor, 2002a, 2002b), and the reaction appears to be dominated by reactive surface-bound species. Furthermore, dissolved ferrous iron can be scavenged by reduced sulfur species (sulfides) to form insoluble precipitates, such as FeS and FeS2. These latter species are formed during sulfate-reducing conditions—which can also result in the reduction of Fe(III) to Fe(II)—and are known as acid volatile sulfides (AVS) or chromium extractable sulfide (CES). Certain AVS and CES species are active in promoting reductive dechlorination during anoxic iron oxidation.
Therefore, only a portion of the total Fe(II) present in soil is chemically reactive because of the requirement for surface interactions with the aqueous medium. Measurements of chemically reactive ferric iron must incorporate mineral phase Fe(II) and should be quantified based on the mass of Fe(II) present per mass of soil. Determining the amount of Fe(II) in a soil matrix that can be used for anoxic iron oxidation during abiotic reductive dechlorination follows the AMIBA (Aqueous and Mineralogical Intrinsic Bioremediation Assessment) protocol (Kennedy et al., 2003).
6.4 Representing Sources in Groundwater Modeling
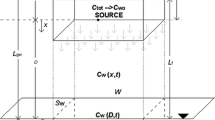
Groundwater transport models use several different methods to simulate the entry of contaminants into groundwater. For example, some analytical models (described below) require that a vertical plane be used as the source of dissolved concentration to the model. The width and depth of the plane in the saturated zone must be entered into the model, followed by the concentration (in units of mass per volume) leaving the plane. This type of source term assumes that groundwater flows through the vertical plane and picks up dissolved phase contamination that then forms the groundwater plume. Other models require that a dissolved mass vs. time function (in units of mass per time) be entered into source zones. Many numerical models use this type of source term to introduce contaminant mass into a simulated groundwater flow field.
The key point is that most models require that the source term be simplified and entered into the groundwater transport model as a relatively abstract source function. Few commonly used groundwater transport models represent the source term directly as a deterministic function of key source processes, such as DNAPL dissolution, leaching of vadose zone materials or matrix diffusion. Users of these groundwater transport models must simplify and develop a source function outside of the model in order to build a source zone in the model (Figure 6.5). In some cases, a constant source zone (unchanging over time) is used to represent a worst-case scenario of plume growth at a site. In other cases, detailed source functions are developed to represent natural weathering of the source, multiple sourcing events and/or removal actions.

Describing the source term in a typical groundwater transport model.
Christ et al. (2006) describe how a number of researchers have developed screening-level models that simulate the change in downgradient groundwater concentrations and source mass over time. It is very difficult with our current state of knowledge to use deterministic models to simulate the changes in source concentrations over time and their associated effects on groundwater concentrations. DNAPL distribution is usually poorly characterized and computational resources are typically constrained (for example, long run-times are undesirable). Thus, having screening models allows an evaluation of source effects without detailed source characterization and with limited computer resources. Christ et al. (2006) evaluated four different screening models—Dekker (1996), Parker and Park (2004), Zhu and Sykes (2004), and Falta et al. (2005a)—all of which employed mass transfer correlations of the following form:
where Keff = upscaled mass transfer coefficient, Ko and \(\beta\) are fitting parameters, M(t) = DNAPL mass at time t and M0 = DNAPL mass at time 0.
The Dekker (1996) model uses a nonsteady, numerically based approach, while the Zhu and Sykes (2004) and the Falta et al. (2005a) models assume simple relationships between mass discharge and mass removal.
The sections below describe some of the key source zone processes and how they can be reduced to source functions for use in groundwater transport models.
6.4.1 DNAPL Sources Zones
The National Research Council (NRC) report (NRC, 2005) on source zone assessment and remediation defines a chlorinated solvent source zone as a subsurface reservoir that sustains a plume. The NRC states that the DNAPL-containing region is typically the primary reservoir but also recognizes that DNAPL mass can be transferred to other source compartments, such as contaminants sorbed to the aquifer matrix or dissolved into low-permeability zones. These other compartments will then function as source zones to groundwater.
DNAPL source zones can be represented by a continuum of sourcing processes that start with an initial DNAPL release that forms DNAPL pools and fingers in the subsurface. In a few cases, when the mass of DNAPL released is large enough and significant capillary barriers (such as unfractured clay layers) are present, free-phase (continuous) DNAPL can accumulate. Because the saturation (the percent of pore space containing DNAPL) is high, this DNAPL can migrate if pumped or if the underlying capillary barrier is disturbed. Most of the time, however, DNAPL is present in source zones as residual DNAPL or as ganglia of DNAPL in a few pores, instead of one large continuous DNAPL mass.
Shortly after release, the DNAPL present in pools and fingers begins to undergo dissolution and to contribute mass to the groundwater system. DNAPL in unsaturated source zones will be exposed to infiltration of precipitation through the vadose zone, forming a leachate that can then enter the saturated zone. DNAPL that migrates to the saturated zone will begin to dissolve immediately as groundwater moves through the DNAPL source zone.
The source function, also called the source zone loading rate or the mass flux from the source zone, can be very difficult to predict with typical site characterization data. The site-specific mix of pools, fingers and source loadings from other source zone compartments at a site is called the source zone architecture. Because the presence of the DNAPL source materials in these zones can be distributed widely over a very sparse pattern (low average DNAPL saturations) that is controlled by micro-stratigraphic features (very small-scale changes in the texture of the water-bearing unit material), detailed information about the source zone architecture is very difficult to obtain. This complexity is illustrated by the phenomenon that at many DNAPL release sites, actual DNAPL is never encountered during the site characterization process.
The importance of understanding the source zone architecture is linked to its ability to control the source function. For example, the individual source functions of DNAPL pools and DNAPL fingers and other source compartments (such as matrix storage) can be very different. A source zone dominated by DNAPL pools, for instance, may experience only minor changes in concentration and mass flux (also called mass discharge, in units of mass per time), leaving the source zone over time, even if significant mass has been dissolved into the downgradient plume. In contrast, a source zone dominated by DNAPL fingers can show significant changes in mass flux even if relatively little mass has been dissolved from the source zone.
Several researchers have proposed that a power function can be used to relate the change in average source concentration (or mass flux) over time to the change in contaminant mass over time (Rao et al., 2001; Rao and Jawitz, 2003; Parker and Park, 2004; Zhu and Sykes, 2004) (see Figure 6.6). This allows the mass discharge (mass flux) to be related to the source mass mathematically at any time. The power function model is given by:

Reduction in mass discharge (mass flux) (%) vs. reduction in source mass (%). Adapted from Stroo et al. (2003) where Curve 1 (top curve) is the theoretical relationship from Rao and Jawitz (2003), Curve 2 is the theoretical relationship from Enfield et al. (2002), Curve 3 is from Newell and Adamson (2005), Curve 4 is actual field data from Rao et al. (1997), and Curve 5 is the theoretical relationship from Sale and McWhorter (2001).
where C(t) = flux-weighted concentration at time t, C0 = flux-weighted concentration at time 0, M(t) = source mass at time t, M0 = source mass at time t0, and \(\Gamma\)= empirical parameter.
On a very simple basis, sources dominated by DNAPL pools will demonstrate a source function below the 45° line on Figure 6.6, while a source dominated by DNAPL fingers will exhibit a source function above the 45° line. Rao and Jawitz (2003) developed a more detailed description by defining “heterogeneous DNAPL distribution in homogenous media” as causing source functions that fall above the line, while “homogeneous DNAPL distribution in heterogeneous media” will have below-the-line source functions. In practice, source zones at actual sites appear to fall both below and above this line (Stroo et al., 2003; Falta et al., 2005a; McGuire et al., 2006) and a middle-of-the-road approach (the 45° line) is a good starting point for selecting a source function if the source architecture is not known.
Many modeling studies have been performed to evaluate the effects of source remediation on the plume. In theory, the source architecture dominates how the source will respond to partial mass removal in two key metrics: mass flux leaving the source zone, and source longevity. For example, removing half of the mass from the upgradient portion of a DNAPL pool can result in the scenario where the mass flux is not changed by any appreciable amount, but the lifetime of the DNAPL pool (the time it takes for the pool to totally dissolve) will be reduced by half. On the other hand, if a source removal technology evenly removes half of the DNAPL residual ganglia throughout a source zone, the resulting mass flux will drop by half. The overall time until the last ganglia dissolve will not change, however. In practice, source removal technologies do appear to be able to change source zone concentrations significantly. One study of 59 source treatment sites showed an average of over 80% reduction in source zone concentrations due to the effects of source treatment technologies, such as enhanced bioremediation, chemical oxidation and thermal treatments (McGuire et al., 2006).
As indicated above, it is currently difficult to translate site characterization data into useable information about source architecture. One approach to address this issue is to use simple mathematical functions to represent the source function over time. Newell and Adamson (2005) studied four simple potential source functions: step function, linear function, first-order decay and a compound model (a plateau function followed by first-order decay). They concluded that because of the continuing overlay of various source decay processes over time in a source zone, such as DNAPL fingers dissolving first, followed by complete dissolution of any longer-lived DNAPL pools, then long-term depletion of contaminants in storage via matrix diffusion, and then slow release of contaminants via dual-equilibrium-desorption (availability) effects, that source functions at most sites will be dominated by long “tails”. Therefore, the use of a first-order decay model, or a companion compound model, was considered to be the most appropriate method to approximate the long-term source function over time (Figure 6.7). Inherent in the use of the planning-level models is that the actual source function will not track the first-order decay model exactly but will exhibit scatter such that different rates of decay over time will be observed. Even so, a first-order or a compound model will capture the essence of a real source function where average concentration and mass flux decline over time with a long-term “tail” over the lifetime of the source.

Two planning level source functions that simulate the expected “tails” caused by source zone architecture: (a) first-order decay and (b) compound model. The source function tail approximates the effects of various processes such as DNAPL dissolution, matrix diffusion, complex desorption processes, and stagnant zones over time (from Newell and Adamson, 2005; reprinted with permission of John Wiley & Sons, Inc).
There are a number of focused research projects in progress at this time that are attempting to make the development of source functions more deterministic (Table 6.3), where site characterization data can lead to definitive and quantitative descriptions of DNAPL source zone architecture and the resulting source function. If successful, groundwater modelers will be able to use this research to develop more robust and accurate source functions for their models.
6.4.2 Estimating Source Mass
Building source functions often requires some estimate of source mass. Considerable research has been devoted to finding better ways to characterize DNAPL source zones (Table 6.3). Even with advanced source characterization methods, there will be considerable uncertainty in source mass estimation.
One key concept in dealing with source mass estimation is to employ the Observational Approach or some type of adaptive site management (such as the Triad approach). With these approaches, the conceptual site model is continually updated with new information. If a groundwater model is built with a source function that assumes a certain mass and the actual source function begins to stray from the predicted source function, then some type of mid-course correction is needed.
Under the Observational Approach (Terzaghi and Peck, 1948; Peck, 1969; NRC, 2006), the following steps are taken:
-
Assess probable conditions and develop contingency plans for adverse outcomes
-
Establish key parameters for observation
-
Measure observational parameters and compare to calculations
-
Compare predicted and measured parameters, and
-
Change the design as needed.
This type of adaptive approach is one method to account for uncertainties in estimating source mass.
Tools have been developed to assist in the calculation of source mass. For example, the SourceDK model (Farhat et al., 2004) has source mass calculation tools for estimating source mass based on sampling data.
6.5 Developed Models
Groundwater transport models are typically divided into two groups: analytical models (based on equations) and numerical models (based on discretization of the model domain and numerical solution methods). In general, analytical models are easier to use and apply, while numerical models are more powerful in their ability to represent detailed site features.
6.5.1 Commonly Used Analytical Models
Several commonly used analytical groundwater models that address chlorinated solvent plumes are presented below. It is noted that some of the models discussed are not technically “analytical models” but are modeling tools that rely on analytical solutions in their development.
6.5.1.1 Biochlor
As described by the USEPA, BIOCHLOR is a screening model that simulates remediation by natural attenuation of dissolved solvents at chlorinated solvent release sites (USEPA, 2008; Aziz et al., 2000a). The model can be used to simulate 1-D advection, 3-D dispersion, linear adsorption and biotransformation via reductive dechlorination (the dominant biotransformation process at most chlorinated solvent sites). Reductive dechlorination of dissolved mass is assumed to occur under anaerobic conditions and to follow a sequential first-order decay process. BIOCHLOR includes three different model types:
-
Solute transport without decay.
-
Solute transport with biotransformation modeled as a sequential first-order decay process.
-
Solute transport with biotransformation modeled as a sequential first-order decay process with two different reaction zones (i.e., each zone has a different set of rate coefficient values).
BIOCHLOR includes a decaying source term where the mass flux and the estimated mass were entered into the model (directly for source mass, indirectly via source zone characteristics for mass flux). The model takes the mass flux and divides it by the source mass to get a first-order source zone decay rate. With this option, the source can be simulated as decaying over time in order to demonstrate the impact of the decaying source on the resulting plume.
6.5.1.2 REMChlor
This chlorinated solvent model combines an analytical solution for time-dependent DNAPL source decay and an analytical advection/dispersion model—the Domenico solution—that has been modified to allow for simulation of chlorinated solvent decay chains (Falta et al., 2005a; 2005b). This model can consider independent variations in parent and daughter compound decay rates and yield coefficients in the plume. Multiple reaction zones can be established to simulate the effects of different geochemical environments and remediation schemes. Currently, the authors are enhancing the model with a groundwater-to-indoor air pathway term and a probabilistic engine to incorporate uncertainty analysis. The model has been peer-reviewed by the USEPA and will be distributed via the USEPA’s Center for Subsurface Modeling Support (CSMoS) web page (http://www.epa.gov/ada/csmos/).
6.5.1.3 ART3D
This 3-D analytic reactive transport model simulates homogeneous and constant transport properties (Quezada et al., 2003, 2004). The model includes complex reaction sequences with first-order decay, as well as 3-D dispersion, and can be applied in forward or inverse and stochastic mode. Users can compare model results to data from observation wells at their sites.
6.5.1.4 Natural Attenuation Software (NAS)
This software package includes both analytical and numerical models that can be used to compare the cleanup times associated with monitored natural attenuation (MNA) to active remediation (Chapelle et al., 2003; Widdowson et al., 2004). Natural attenuation processes modeled by NAS include advection, dispersion, sorption, nonaqueous phase liquid (NAPL) dissolution and biodegradation. NAS determines redox zonation and estimates and applies varied biodegradation rates from one redox zone to the next. NAS calculates three key MNA variables:
-
Required Source Reduction: target source concentration required for a plume extent to contract to regulatory limits (Distance of Stabilization [DOS])
-
Time of Stabilization (TOS): time required for a plume extent to contract to regulatory limits after source reduction, and
-
Time of Remediation (TOR): time required for NAPL contaminants in the source area to attenuate to a predetermined target source concentration.
6.5.1.5 MNAtoolbox
This software package can be used to screen sites for the potential implementation of MNA (Brady et al., 2001). MNAtoolbox can be used to identify the primary attenuation pathways and to validate the conditions that might prohibit the use of MNA for particular contaminants. Site-specific input parameters are used to gauge the probable effectiveness of attenuation for each contaminant at a given site.
6.5.1.6 BioBalance Toolkit
This software package uses a mass-balance approach to evaluate whether the assimilative capacity of a particular system is sufficient to manage the mass flux of chlorinated solvents emanating from a source zone (Kamath et al., 2006). This modeling platform integrates two planning-level source functions into an analytical model. Users have the option to define the source as either being a vadose zone or submerged source, coupled with the ability to simulate the effects of source management activities by changing the mass flux or the source mass or both. The model includes modules that help estimate the relative abundance of electron donor supply vs. electron donor demand (in units of dissolved hydrogen equivalents), calculate the potential loss of available donor due to the effects of competing electron acceptors, and evaluate plume stability and the relative contribution of various dissolved-phase natural attenuation processes.
6.5.2 Current Numerical Models
A variety of numerical models have been developed to simulate the transport and biotransformation of dissolved chlorinated organic compounds in the subsurface. These include advanced research models capable of simulating multiphase flow, NAPL dissolution and a variety of chemical and biological transformation reactions (Christ et al., 2005). However, the most commonly employed models are based on the groundwater flow model MODFLOW (McDonald and Harbaugh, 1988), and the groundwater solute transport model MT3D (Zheng, 1990). These codes are popular with model developers because of their modular construction, robust numerical methods, good documentation, and continued maintenance and upgrading. These codes are also popular with model users because of the availability of graphical user interfaces (GUIs) including Visual Modflow (http://www.visual-modflow.com), GMS (http://www.ems-i.com), Groundwater Vistas (http://www.groundwater-vistas.com) and Argus ONE (http://www.argusint.com). Contaminant transport and transformation models based on MODFLOW-MT3D include SEAM3D (Waddill and Widdowson, 2000; Widdowson, 2003), BioRedox (Carey et al., 1999; Schreiber et al., 2004), RT3D (Clement, 1997) and PHT3D (Prommer, 2002; Prommer et al., 2003).
6.5.2.1 SEAM3D
SEAM3D is based on the code MT3DMS (Zheng and Wang, 1999) with additional packages for biodegradation via direct oxidation, reductive dechlorination, cometabolism and NAPL dissolution (Waddill and Widdowson, 2000; Widdowson, 2003):
-
The biodegradation package simulates the direct oxidation of organic substrates under a range of terminal electron accepting processes (TEAPs). Biodegradation follows Monod kinetics where the degradation rate is controlled by the concentration of electron donor (ED), electron acceptor (EA), a rate inhibition term and the microbial population. The model may be operated under either growth (time-dependent microbial population) or no-growth (steady-state biomass concentration) conditions. The microbial phase is assumed to include as many as nine different bacterial populations that exist as scattered microcolonies attached to the porous medium. Different TEAPs are simulated in a sequential manner where oxygen is consumed first, followed by nitrate, Mn(IV), Fe(III) and sulfate followed by methanogenic fermentation. cis-1,2-Dichloroethene (cis-DCE) and vinyl chloride (VC) may be degraded via direct oxidation depending on the availability of electron acceptors.
-
The reductive dechlorination package simulates biodegradation of PCE, trichloroethene (TCE), cis-DCE and VC in the absence of oxygen or nitrate (Widdowson, 2003). Reductive dechlorination is assumed to occur at the maximum rate under methanogenic conditions with somewhat slower rates under Mn(IV)-, Fe(III)- and sulfate-reducing conditions where the current TEAP for each cell is determined by the biodegradation package. Chlorinated ethene degradation rates are assumed to decrease in the order PCE→TCE→cis-DCE→VC.
-
The cometabolism package is designed to simulate aerobic cometabolism of user-designated compounds (recalcitrants) using methane or petroleum-derived compounds (e.g., toluene) as co-substrates. Recalcitrants may be TCE, cis-DCE or VC or any user-defined compound (e.g., methyl tertiary butyl ether [MTBE]).
-
The NAPL dissolution package can be used to simulate the dissolution of a multi-component NAPL, where the rate of mass transfer between the NAPL and groundwater is a function of the interfacial mass transfer rate and the difference between the aqueous phase and the equilibrium concentration. For multi-component NAPLs, the equilibrium concentration is calculated using Raoult’s Law.
SEAM3D is a powerful modeling program, allowing simulation of a wide variety of biogeochemical processes. However, this versatility also increases the model complexity and the number of input parameters. For many of the microbial processes, there are no published values of input parameters or methods for independently estimating these parameters. Without accurate estimates of all of the important parameters, it is very difficult to evaluate the accuracy of any model simulation.
6.5.2.2 BioRedox
BioRedox-MT3DMS (BioRedox) is a 3-D, multicomponent solute transport model that was developed to simulate the natural and enhanced bioremediation of chlorinated solvents and petroleum hydrocarbons in groundwater (Carey et al., 1999; Schreiber et al., 2004). BioRedox is based on MT3DMS DoD_3.00.A (Zheng and Wang, 1999) and includes a reaction module and a multicomponent DNAPL dissolution module that are incorporated into the MT3DMS source code.
BioRedox provides the option to simulate conditions that range from a simple scenario with first-order decay for one species to more sophisticated conditions that include the coupled oxidation-reduction of multiple electron donors and electron acceptors. The strength of BioRedox is the flexibility incorporated into the reaction and DNAPL dissolution modules, which allows users to simulate a broad range of site-specific conditions without having to modify the source code.
BioRedox functionality can include simulation of the following processes at a site:
-
Biodegradation mechanisms such as oxidation, reductive dechlorination and cometabolism
-
Representation of aqueous and/or mineral species
-
Oxidation of organic compounds (e.g., benzene, toluene, ethylbenzene and/or xylene [BTEX]) coupled to the reduction of inorganic electron acceptors (e.g., oxygen, nitrate, mineral-phase manganese and iron, sulfate and/or carbon dioxide)
-
A unique visualization technique that allows users to simultaneously evaluate the effects of simulated redox zones on contaminant distributions that are based on TEAP-specific processes
-
Simulation of halogen accumulation (e.g., chloride production during the reductive transformation of chlorinated solvent species)
-
Production of dissolved species such as ferrous iron during the reduction of mineral-phase ferric iron, or the production of methane in the methanogenic zone (BioRedox can assume unlimited carbon dioxide availability so that this species does not have to be represented explicitly in a site model)
-
Biodegradation mechanisms, rates and daughter products can vary in each simulated redox zone (e.g., vinyl chloride oxidation in aerobic and iron-reducing zones, and reductive dechlorination in the methanogenic zone)
-
Accelerated biodegradation rates that occur during methanotrophic cometabolism (e.g., rapid biodegradation of TCE under aerobic condition when simulated substrates such as methane or toluene exceed a user-defined threshold concentration)
-
Reaction rates that are first-order, instantaneous (e.g., BTEX or ferrous iron oxidation under aerobic conditions), or substrate-limited reaction rates
-
Representation of multicomponent inhibition or stimulation by allowing users to specify two biodegradation rates for a given redox zone: one rate that applies when a co-substrate or an inhibitor is present within a user-defined range of threshold concentrations, and a different rate that applies when the co-substrate or inhibitor is outside the range of threshold concentrations, and
-
Multicomponent DNAPL dissolution simulation by calculating the effective solubility for each component and either equilibrium or rate-limited dissolution.
BioRedox is capable of simulating many of the same microbial processes as SEAM3D. However, because BioRedox allows for more simplistic representations of biodegradation processes (including first-order decay and instantaneous reaction kinetics), fewer input parameters may be required, allowing the use of these approaches when calibration data are more limited.
6.5.2.3 RT3D
RT3D (Reactive Transport in 3-Dimensions; Clement, 1997) is also based on the MODFLOW and MT3D codes and is designed to allow users to simulate a variety of processes including oxidative biodegradation, reductive dechlorination and NAPL dissolution. However, RT3D is unique in that it couples the solute transport components of MT3D with an implicit ordinary differential equation solver (Hindmarsh, 1983) allowing the code to be used to simulate a wide variety of chemical reactions. RT3D users can define their own reaction packages in order to adapt the numerical model to a site-specific conceptual model. Consequently, RT3D can be easily adapted as remediation technologies evolve and as the understanding of in situ transformation processes improves. This potential is best illustrated by evolution of the RT3D model.
The basic structure of the RT3D model (RT3D v1.0) has remained unchanged since it was first released (Clement, 1997) with seven pre-programmed reaction modules to simulate (1) instantaneous substrate oxidation using oxygen, (2) instantaneous substrate oxidation using multiple electron acceptors, (3) first-order kinetic substrate oxidation using multiple electron acceptors, (4) rate-limited sorption, (5) double Monod kinetics of substrate oxidation using a single electron acceptor, (6) first-order sequential decay of chlorinated ethenes and (7) aerobic/anaerobic biodegradation of chlorinated ethenes. Later, reaction modules were released to simulate NAPL dissolution (Clement et al., 2004) and mixtures of chlorinated ethenes, ethanes, methanes and their daughter products (Johnson and Truex, 2006). Coulibaly et al. (2006) and Jung et al. (2006) recently employed the user-defined module feature in RT3D to simulate emulsified oil transport for chlorinated solvent bioremediation.
The ability to select and/or develop reaction modules to match site-specific conditions is one of the great strengths of RT3D, allowing the model to be used under a variety of conditions including relatively simple sites (e.g., single contaminant and redox condition) with limited input data and highly complex sites (e.g., multiple contaminants degrading under a variety of redox conditions) with much more extensive input data.
6.5.2.4 PHT3D
A variety of coupled transport and geochemical models have been developed to simulate solute transport under conditions where the chemical reactions are fast relative to groundwater flow. In this case, a geochemical equilibrium approach is used to determine the fraction of each species in the mobile, aqueous phase (e.g., Yeh and Tripathi, 1989; Steefel and MacQuarrie, 1996). While this approach may be appropriate for certain reactions (e.g., ion exchange), it is not appropriate for many contaminant biodegradation reactions where reaction kinetics can be much slower than groundwater flow.
PHT3D (Prommer, 2002; Prommer et al., 2003) represents an attempt to merge some of the attractive features of the multi-species biodegradation models (e.g., RT3D) with geochemical models. PHT3D (v1.0) can handle general mixed equilibrium/kinetic geochemical reactions by linking the MODFLOW/BioRedox-MT3DMS flow/transport models with the non-equilibrium geochemical model PHREEQC-2 (Parkhurst and Appelo, 1999). Because the reaction part of the model is based on PHREEQC-2, reaction kinetics easily can be formulated through user-defined rate expressions within the database.
PHT3D will be most useful in situations where complex interactions between organic and inorganic species must be considered. Examples of this type of problem include abiotic degradation of chlorinated solvents in permeable reactive barriers and enhanced anaerobic biodegradation where mobilization of solid-phase species (e.g., Mn, Fe, As) is a concern. PHT3D typically requires a large amount of input data for model calibration. However, much of the geochemical data is already available within the PHREEQC-2 (Parkhurst and Appelo, 1999) and other readily available databases.
6.6 Case Studies
6.6.1 BIOCHLOR Case Study—Cape Canaveral Air Station, Fire Training Area, Florida
Aziz et al. (2000a) applied the BIOCHLOR model to simulate the movement of chlorinated solvents and determine the percent of TCE biotransformation at Cape Canaveral Air Station, Florida. The study focused on a former fire training area on the western edge of the station, about 335 meters (m), or 1,100 feet (ft) east of the Banana River and about 305 m (1,000 ft) northeast of a canal that empties into the river. Fire-training activities at the site from about 1965 to 1985 resulted in contamination of shallow soils and groundwater with a mixture of chlorinated solvents and fuel hydrocarbons. Other constituents of concern at the site are PCE, cis-DCE and VC.
The upper aquifer at the site consists of about 3 to 4.5 m (10 to 15 ft) of fine to coarse sand and shell fragments with occasional clay lenses and peat stringers (Wiedemeier et al., 1999). This unit is underlain by the Caloosahatchee Marl, consisting of fine-grained calcareous sand, unconsolidated shells and shell fragments, with some interbedded clay units and peat stringers. About 15 to 18 m (50 to 60 ft) below ground surface (bgs), a clay layer is present within the Marl.
Depth to groundwater at the site ranges from about 0.7 m (2 ft) bgs in wells near surface-water bodies to about 3.4 m (11 ft) bgs in wells northeast of the site (Wiedemeier et al., 1999). The hydraulic gradient is approximately 0.0012 m/m, based on observed static water level measurements, with groundwater discharging to the canal and the Banana River. The hydraulic conductivity for the unconfined aquifer averages about 1.8 centimeters per second (cm/sec) based on slug test results. The effective porosity is assumed to be 0.20.
Prior to determining the amount of TCE biotransformation at the site in 1998, BIOCHLOR was calibrated to match site conditions. For this purpose, the model was used to reproduce the movement of the plume from the best guess for when the release occurred (1965) to 1997 (the data available to Aziz et al. [2000a] for model calibration).
The aquifer was estimated to have a longitudinal dispersivity of 12.2 m (40 ft), based on the intermediate value for a plume length of 243–366 m (800–1,200 ft) (Gelhar et al., 1992). A transverse dispersivity of 1.3 m (4 ft) or 10% of the longitudinal dispersivity, was assumed. A vertical dispersivity of zero was assumed since the depth of the source was approximately equal to the depth of the aquifer.
6.6.1.1 Contaminant Retardation Factors
Contaminant retardation factors were calculated using:
where:
-
R = coefficient of retardation factor (unitless)
-
Koc = soil sorption coefficient (liters/kilogram [L/kg])
-
foc = fraction organic carbon (kg organic carbon/kg soil)
-
ρb = aquifer bulk density (kg/L)
-
n = porosity (L/L)
Individual retardation factors of PCE = 7.1, TCE = 2.9, cis-DCE = 2.8, VC = 1.4, and ethene = 5.3 were obtained using:
-
An estimated aquifer bulk density of 1.6 kg/L
-
foc of 0.184% based on a laboratory analysis of site aquifer solids
-
Koc values of PCE = 426 L/kg, TCE = 130 L/kg, cis-DCE = 125 L/kg, VC = 29.6 L/kg, and ethene = 302 L/kg using literature correlations of solubilities at 20 degrees Celsius (°C), and
-
An effective porosity (n) of 0.20.
However, BIOCHLOR uses one retardation factor, not individual retardation factors for each constituent. For this study, the median value of 2.85 was chosen.
Because the case study focused on the percent of TCE biotransformation, the source area was modeled as a spatially variable source. To obtain the most conservative centerline predictions, the maximum concentrations in the source area were used for the first zone. Concentrations for the other two zones were obtained by taking the geometric means between adjacent isopleths (see Figure 6.8).

TCE site characterization data for Cape Canaveral Air Station, Fire Training Area, Florida for input to BIOCHLOR (from Aziz et al., 2000a).
A source thickness of 17 m (56 ft) was based on the deepest depth where chlorinated solvents were detected in the aqueous phase. A modeled area length of 330 m (1,085 ft) was calculated as the distance from the source to the receptor (the canal, in this case study). A source width of 213 m (700 ft), significantly larger than the plume width, was used to capture all of the mass discharging into the canal. A simulation time of 33 years was used since the study focused on calculating the percent biotransformation in 1998 and the solvents were released starting in 1965.
Contaminant biotransformation was modeled as a single anaerobic zone based on field dissolved oxygen, oxidation-reduction potential (ORP), and geochemical data that were used to establish anaerobic conditions. Because field-scale rate coefficients and rate data from microcosms were unavailable, literature values were used as the starting rate coefficients. Final rate coefficients were obtained by calibrating the model to field data.
Once the boundary and initial conditions had been defined, the BIOCHLOR model was calibrated by adjusting the biotransformation rate coefficients until the best fit to 1997 field data was obtained as shown in Figures 6.9 to 6.11. Final biotransformation rate coefficients obtained were: PCE→TCE = 2.0 yr-1; TCE→cis-DCE = 1.0 yr-1; cis-DCE→VC = 0.7 yr-1; and VC→ethene (ETH) = 0.4 yr-1.

TCE centerline concentration output data for Cape Canaveral Air Station, Fire Training Area, Florida (from Aziz et al., 2000a).

cis-DCE centerline concentration output data for Cape Canaveral Air Station, Fire Training Area, Florida (from Aziz et al., 2000a).

VC centerline concentration output data for Cape Canaveral Air Station, Fire Training Area, Florida (from Aziz et al., 2000a).
Once the model had been calibrated, the percent of TCE biotransformation was calculated as approximately 84% (see Figure 6.12).

Array concentration output data for TCE at Cape Canaveral Air Station, Fire Training Area, Florida (from Aziz et al., 2000a).
6.6.2 RT3D Case Study—Simulation of an Emulsified Oil Biobarrier Treating Chlorinated Solvents and Perchlorate
Borden (2007a) applied the numerical model RT3D to simulate the transport and enhanced anaerobic biodegradation of chlorinated solvents and perchlorate in an emulsified oil biobarrier. The biobarrier was installed in a shallow unconfined aquifer impacted by ammonium perchlorate and waste solvent released from a former surface impoundment. The primary COCs at the pilot-test site were perchlorate (ClO- 4) and 1,1,1-trichloroethane (1,1,1-TCA) with lesser amounts of TCE and related chlorinated volatile organic compounds (CVOCs). The water table aquifer at the site was composed of silty sand and gravel to approximately 5–6 m (16–20 ft) bgs and was underlain by silty clay.
The 15.2 m (50 ft) wide biobarrier was installed in a three-step process. First, 10 direct push wells were installed 1.5 m (5 ft) on center, with 3 m (10 ft) of 2.5 cm (1 inch [in]) diameter #20 slotted polyvinyl chloride (PVC) screen. Over a 5-hour period, every other well was simultaneously injected with 42 L (11 gal) of the pre-blended concentrated emulsion (Edible Oil Substrate or EOS®) diluted with 166 L (44 gallons [gal]) of water, followed by 625 L (165 gal) of chase water to distribute and immobilize the emulsion. The next day, the remaining wells were treated following the same procedure (Borden, 2007b; Zawtocki, 2005).
Contaminant transport and biodegradation in the emulsified oil barrier and downgradient aquifer were simulated using the sequential decay model (Module #6) within RT3D (Clement, 1997) where:
and:
-
A = aqueous phase concentration (moles/L) for compound A
-
RA = retardation factor for compound A
-
D = dispersion coefficient (m/d2)
-
v = velocity (m/d)
-
φ = porosity (dimensionless)
-
qs = volumetric water flux per volume of aquifer representing sources and sinks
-
As = source/sink concentration (moles/L) for contaminant A
-
Soil = sediment oil concentration (g oil/g sediment)
-
kA = effective 2nd order decay coefficient (g sediment/g oil - d) for contaminant A
-
YB/A = degradation product yield coefficient (moles of B produced per mole of A degraded)
The actual numerical simulations were conducted using GMS 6.0 (Brigham Young University, 2005) to aid in data entry and visualization of model output. Prior to simulating contaminant transport, MODFLOW (McDonald and Harbaugh, 1988) was calibrated to match conditions at the field site. The aquifer was represented as a two-layer system (each layer 1.5 m [4.9 ft] thick) with injection and monitor wells fully penetrating both the shallow and deeper layers. The shallow and deeper layers were estimated to have hydraulic conductivities (K) of 2.1 and 10.7 meters/day (m/d) based on visual observations of soil cores, grain size analyses and slug tests.
Flow and transport parameters were estimated by matching model simulations to results from the non-reactive tracer tests (hydraulic gradient = 0.005, effective porosity = 0.18, longitudinal dispersivity = 0.3 m, horizontal transverse dispersivity = 0.03 m, vertical transverse dispersivity = 0.003 m, bulk density = 1.85 g/cm3). Following emulsion injection, K was assumed to be reduced by up to 77% due to clogging with accumulated biomass, gas bubbles and/or retained oil (Borden, 2007a). The reduction in K was assumed to be linearly proportional to retained oil concentration where Kreduced = Kinitial × (1 – 0.77 ×Soil/Maximum Soil). Oil concentration in the sediment (Soil) was determined from numerical simulations of oil droplet transport and retention immediately following emulsion injection and varied from a maximum value of 0.0026 g oil/g sediment adjoining the injection wells to zero in untreated areas (Borden, 2007b).
Contaminant biodegradation was modeled as a second-order process where the contaminant decay rate was assumed to be linearly proportional to both Soil and the contaminant concentration. Soil was determined from prior simulations of oil droplet transport and retention (Borden, 2007b) and assumed constant over the simulation period. The contaminant transport model was first calibrated to match the observed contaminant distribution prior to emulsion injection using specified concentration cells to define the upgradient boundary condition. Contaminant retardation factors were estimated based on published soybean oil-water and octanol-water partition coefficients (Howard, 1990). Long and Borden (2006) found that a linear-equilibrium partitioning approach provided good estimates of the retardation of PCE in abiotic laboratory columns treated with emulsified soybean oil.
Once boundary and initial conditions were defined, the model was calibrated by adjusting the second-order degradation rate (k) for each contaminant until the mean error between observed and simulated steady-state concentrations was less than 0.5% of the average concentration. Estimated second-order degradation rates for each contaminant are shown in Table 6.4. Once calibrated, the model was used to evaluate the effect of alternative barrier designs on chlorinated solvent treatment efficiency.
6.6.2.1 Full-Scale Barrier Performance
Once calibrated, the model was used to evaluate two different barrier alternatives. For both alternatives, a 10 m (33 ft) wide portion of the barrier was simulated with the model domain extending 15 m (49 ft) upgradient of the barrier and 30 m (98 ft) downgradient. Since only a short section of barrier was simulated, upgradient contaminant concentrations were assumed to be uniform and equal to 9,000 micromolar (µM) ClO- 4, 57.4 µM TCA, 0.5 µM DCA and 0.08 µM CA.
For Barrier Alternative I, injection wells were spaced 5 m (16 ft) apart, perpendicular to groundwater flow with continuous screens extending through both the shallow and deeper layers. A 20% EOS: 80% water solution was injected in every other well for 0.5 days, followed by chase water for 1.5 days. This procedure was then repeated with the remaining wells. The emulsion and chase-water injection flow rate was 4 m3/d per well to match the flow rate previously used in the field. The flow rate entering each layer was assumed to be proportional to the layer transmissivity with 16% of the flow entering the shallow, less permeable layer (K = 2.1 m/d) and 84% of the flow entering the deeper, more permeable layer (K = 10.7 m/d).
Figure 6.13 shows the simulated oil distribution in the sediment following injection and the steady-state aqueous contaminant distributions in the shallow, less permeable layer of the aquifer for Barrier Alternative I. In the deeper, more permeable layer, oil was effectively distributed between the injection wells, resulting in a relatively uniform oil-treated zone 13–18 m long along the direction of groundwater flow with a hydraulic retention time (HRT) of 3–4 months (data not shown for the deeper layer). The relatively high residual oil concentrations throughout the deeper layer (0.0037 g/g) combined with the large HRT resulted in very high treatment efficiencies in the deeper layer, with all contaminants reduced below applicable regulatory standards. However, in the shallow layer (shown in Figure 6.13), the oil distribution was much less uniform, varying from 0.0037 g/g near the injection wells to 0.00033 g/g midway between the injection wells. This nonuniform oil distribution allowed substantial amounts of contaminants to pass untreated through the gaps in the oil-treated zone. Farther downgradient, residual contaminants diffused from the shallow layer into the deeper layer.

Model prediction of oil distribution in sediment following injection and steady-state aqueous contaminant distribution for Barrier Alternatives I and II (from Borden, 2007b).
Barrier Alternative II used one set of continuously screened injection wells spaced 5 m apart (same as Alternative I) with a second set of injection wells located midway between the first set and screened only in the shallow, less permeable layer. The injection process for the first set of wells was identical to Alternative I. Once the first set of injections was complete, a 20% EOS: 80% water solution was injected into the second set of wells for 0.5 days, followed by chase water for 1.5 days at a flow rate of 0.66 m3/d per well.
Figure 6.13 shows the simulated oil distribution in the sediment following injection and the steady-state aqueous contaminant distributions in the shallow, less permeable layer for Barrier Alternative II. The modified injection procedure followed in Alternative II resulted in continuous oil-treated zones in both the shallow and deeper layers. The zones of high oil residual saturation (∼ 0.0037 g/g) were 5–6 m long in the shallow layer and 13–17 m long in the deeper layer with HRTs that vary from 4–5 months in the shallow layer to 3–4 months in the deeper layer. The long contact time between the contaminants and oil in both the shallow and deeper layers resulted in very effective treatment, with all contaminants reduced below treatment standards in both the shallow and deeper layers.
6.7 Conclusion and Summary
In summary, and as evidenced by the numerous model development efforts that have been undertaken or are under way for chlorinated solvents, much progress has been made in our ability to articulate and understand the various mechanisms and processes that affect the fate and transport of a chlorinated compound in groundwater. Pilot-scale and large-scale studies at chlorinated solvent sites have greatly assisted the process of model development, application and validation. Additionally, the numerous laboratory- and field-scale biodegradation studies have allowed development of kinetic expressions and biodegradation rate data to support the modeling efforts. Challenges still remain, however, in modeling sources and their behavior over time and in modeling the complexities of the biodegradation process. Research is needed to improve the tools developed to date and their predictive capabilities.
References
Alvarez-Cohen L, Speitel GE. 2001. Kinetics of aerobic metabolism of chlorinated solvents. Biodegradation 12:105–126.
Anderson MP, Woessner WW. 1992. Applied Groundwater Modeling. Academic Press, San Diego, CA, USA. 381 p.
Aziz CE, Newell CJ, Gonzales JR, Haas PE, Clement TP, Sun Y. 2000a. BIOCHLOR Natural Attenuation Decision Support System, User’s Manual Version 1.0 EPA/600/R-00/008. USEPA, Office of Research and Development, Washington, DC, USA. January. http://www.gsi-net.com. Accessed June 9, 2009.
Aziz CE, Smith AP, Newell CJ, Gonzales JR. 2000b. BIOCHLOR Chlorinated Solvent Plume Database Report. Prepared for the Air Force Center for Environmental Excellence, Brooks City-Base, TX, USA. June. http://www.afcee.af.mil/resources/technologytransfer/models/index.asp. Accessed June 9, 2009.
Bear J, Verrujit A. 1987. Modeling Groundwater Flow and Pollution. D. Reidel, Dordrecht, Holland. 414 p.
Bedient PB, Rifai HS, Newell CJ. 1999. Ground Water Contamination: Transport and Remediation, 2nd Edition. Prentice-Hall, Upper Saddle River, NJ, USA. 604 p.
Borden RC. 2007a. Concurrent bioremediation of perchlorate and 1,1,1–trichloroethane in an emulsified oil barrier. J Contam Hydrol 94:13–33.
Borden, RC. 2007b. Effective distribution of emulsified edible oil for enhanced anaerobic bioremediation. J Contam Hydrol 94:1–12.
Borden RC, Bedient PB. 1986. Transport of dissolved hydrocarbons influenced by oxygen-limited biodegradation: 2. Field application. Water Resour Res 22:1983–1990.
Bouwer EJ, McCarty PL. 1983. Transformations of 1– and 2–carbon halogenated aliphatic organic compounds under methanogenic conditions. Appl Environ Microbiol 45:1286–1294.
Brady PV, Spalding BP, Krupka KM, Waters RD, Zhang P, Borns DJ. 2001. Site Screening for Monitored Natural Attenuation with MNAtoolbox. Sandia National Laboratories. http://www.sandia.gov/eesector/gs/gc/na/mnahome.html. Accessed June 9, 2009.
Bredehoeft J. 1994. The conceptualization model problem–surprise. Hydrogeol J 13:37–46.
Brigham Young University. 2005. The Department of Defense Groundwater Modeling System, GMS v6.0, Environmental Modeling Research Laboratory. http://www.emrl.byu.edu/gms.htm. Accessed June 9, 2009.
Buscheck TE, Alcantar CM. 1995. Regression techniques and analytical solutions to demonstrate intrinsic bioremediation. In Hinchee RE, Wilson JT, Downey DC, eds, Intrinsic Bioremediation: Proceedings of the Third International In Situ and Onsite Bioremediation Symposium, Vol 3(1). Battelle Press, Columbus, OH, USA, pp 109–116.
Carey GR, Van Geel PJ, Murphy JR. 1999. BIOREDOX MT3DMS: A coupled biodegradation redox model for simulating natural and enhanced bioremediation of organic pollutants – V2.0 User’s Guide. Conestoga Rovers & Associates, Waterloo, Ontario, Canada.
Carrera J, Alcolea A, Median A, Hidalgo J, Sllot A. 2005. Inverse problem in hydrogeology. Hydrogeol J 13:206–222.
Chapelle FH, Bradley PM, Lovley DR, Vroblesky DA. 1996. Measuring rates of biodegradation in a contaminated aquifer using field and laboratory methods. Ground Water 34:691–698.
Chapelle FH, Widdowson MA, Brauner JS, Mendez E, Casey CC. 2003. Methodology for estimating times of remediation associated with monitored natural attenuation. U.S. Geological Survey Water-Resources Investigations Report 03–4057. 51 p.
Chen W, Kan AT, Newell CJ, Moore E, Tomson MB. 2002. More realistic soil cleanup standards with dual-equilibrium desorption. Ground Water 40:153–164.
Chen W, Lakshmanan K, Kan AT, Tomson MB. 2004. A program for evaluating dual-equilibrium desorption effects on remediation. Ground Water 42:620–624.
Christ JA, Ramsburg CA, Abriola LM, Pennell KD, Löffler FE. 2005. Coupling aggressive mass removal with microbial reductive dechlorination for remediation of DNAPL source zones: A review and assessment. Environ Health Perspectives 113:465–477.
Christ JA, Ramsburg CA, Pennell KD, Abriola LM. 2006. Estimating mass discharge from dense nonaqueous phase liquid source zones using upscaled mass transfer coefficients: An evaluation using multiphase numerical simulations. Water Resour Res 42:W11420.
Clement TP. 1997. A Modular Computer Code for Simulating Reactive Multispecies Transport in 3–Dimensional Groundwater Aquifers. Technical Report PNNL-SA-28967. Pacific Northwest National Laboratory, Richland, WA, USA.
Clement TP, Gautam TR, Lee KK, Truex MJ, Davis GB. 2004. Modeling of DNAPL-dissolution, rate-limited sorption and biodegradation reactions in groundwater systems. Bioremediation J 8:47–64.
Cohen RM, Mercer JW. 1993. DNAPL Site Evaluation. CRC Press, Inc., Boca Raton, FL, USA. 384 p.
Coulibaly KM, Long CM, Borden RC. 2006. Transport of edible oil emulsions in clayey-sands: 1–D column results and model development. J Hydrol Eng 11:230–237.
Dekker TJ. 1996. An assessment of the effects of field-scale formation heterogeneity on surfactant-enhanced aquifer remediation. PhD Dissertation, University of Michigan, Ann Arbor, MI, USA.
Doherty J. 2003. Ground water model calibration using pilot points and regularization. Ground Water 41:170–177.
Doherty, J. 2005. PEST: Software for Model-Independent Parameter Estimation. Watermark Numerical Computing, Australia.
Domenico PA. 1987. An analytical model for multidimensional transport of a decaying contaminant species. J Hydrol 91:49–58.
Domenico PA, Schwartz FW. 1998. Physical and Chemical Hydrogeology, 2nd ed. John Wiley and Sons, New York, NY, USA. 528 p.
Ely RL, Williamson KJ, Hyman MR, Arp DJ. 1997. Cometabolism of chlorinated solvents by nitrifying bacteria: Kinetics, substrate interactions, toxicity effects, and bacterial response. Biotechnol Bioeng 54:520–534.
Enfield CG, Wood AL, Brooks MC, Annable MD. 2002. Interpreting tracer data to forecast remedial performance. In Thornton S, Oswald S, eds, Groundwater Quality: Natural and Enhanced Restoration of Groundwater Pollution. Publication No. 275. International Association of Hydrologic Sciences Press, Wallingford, Oxfordshire, UK, pp 11–16.
Falta RW, Rao PSC, Basu N. 2005a. Assessing the impacts of partial mass depletion in DNAPL source zones: I. Analytical modeling of source strength functions and plume response. J Contam Hydrol 78:259–280.
Falta RW, Basu N, Rao PSC. 2005b. Assessing the impacts of partial mass depletion in DNAPL source zones: II. Coupling source strength functions to plume evolution. J Contam Hydrol 79:45–66.
Farhat SK, de Blanc PC, Newell CJ, Gonzales JR, Perez J. 2004. SourceDK Remediation Timeframe Decision Support System, User’s Manual. Prepared for the Air Force Center for Environmental Excellence, Brooks City-Base, TX, USA. http://www.gsi-net.com. Accessed June 9, 2009.
Ferrey ML, Wilkin RT, Ford RG, Wilson JT. 2004. Nonbiological removal of cis-dichloroethylene and 1,1–dichloroethylene in aquifer sediment containing magnetite. Environ Sci Technol 38:1746–1752.
Fogel MM, Taddeo AR, Fogel SS. 1986. Biodegradation of chlorinated ethenes by a methane-utilizing mixed culture. Appl Environ Microbiol 51:720–724.
Folsom BR, Chapman PJ, Pritchard PH. 1990. Phenol and trichloroethylene degradation by Pseudomonas Cepacia G4: Kinetics and interactions between substrates. Appl Environ Microbiol 56:1279–1285.
Freyberg DL. 1988. An exercise in ground-water model calibration and prediction. Ground Water 26:350–360.
Gelhar LW, Welty C, Rehfeldt KR. 1992. A critical review of data on field scale dispersion in aquifers. Water Resour Res 28:1955–1974.
Guyonnet D, Neville C. 2004. Dimensionless analysis of two analytical solutions for 3–D solute transport in groundwater. J Contam Hydrol 75:141–153.
Hausman S, Rifai HS. 2005. Modeling remediation time using natural attenuation at a dry cleaner site. Remediat J 16:5–31.
Hindmarsh AC. 1983. ODEPACK, a systemized collection of ODE solvers. In Stepleman RS, ed, Scientific Computing, Amsterdam, Holland. 55 p.
Hirschorn SK, Grostern A, Lacrampe-Coulome G, Edwards EA, MacKinnon L, Repta C, Major DW, Sherwood Lollar B. 2007. Quantification of biodegradation of chlorinated hydrocarbons: Added value via stable carbon isotope analysis. J Contam Hydrol 94:249–260.
Hopkins GD, McCarty PL. 1995. Field evaluation of in situ aerobic cometabolism of trichloroethylene and three dichloroethylene isomers using phenol and toluene as the primary substrates. Environ Sci and Technol 29:1628–1637.
Howard PH. 1990. Handbook of Environmental Fate and Exposure Data for Organic Chemicals. Volume II, Solvents. Lewis Publishers, Chelsea, MI, USA. 576 p.
Hunt RJ, Doherty J, Tonkin MJ. 2007. Are models too simple? Arguments for increased parameterization. Ground Water 45:254–262.
Johnson CD, Truex MJ. 2006. RT3D Reaction Modules for Natural and Enhanced Attenuation of Chloroethanes, Chloroethenes, Chloromethanes, and Daughter Products. Technical Report PNNL-15938, Pacific Northwest National Laboratory, Richland, WA, USA.
Jung Y, Coulibaly KM, Borden RC. 2006. Transport of edible oil emulsions in clayey-sands: 3–D sandbox results and model validation. J Hydrol Eng 11:238–244.
Kamath RK, Newell CJ, Looney BB, Vangelas KM, Adamson DT. 2006. BioBalance–A Mass Balance Toolkit, User’s Manual. May. http://www.gsi-net.com. Accessed June 9, 2009.
Kelson VA, Hunt RJ, Haitjema HM. 2002. Improving a regional model using reduced complexity and parameter estimation. Ground Water 40:132–143.
Kennedy L, Everett J, Gonzales J. 2003. AMIBA (Aqueous and Mineralogical Intrinsic Bioremediation Assessment) Protocol. Air Force Center for Environmental Excellence, Brooks City-Base, TX, USA. http://www.afcee.af.mil/shared/media/document/AFD-071211-022.pdf. Accessed June 9, 2009.
Konikow LF, Bredehoeft JD. 1992. Ground-water models cannot be validated. Adv in Water Resour 15:75–83.
Lee W, Batchelor B. 2002a. Abiotic reductive dechlorination of chlorinated ethylenes by iron bearing soil minerals: 1. Pyrite and magnetite. Environ Sci Technol 36:5147–5154.
Lee W, Batchelor B. 2002b. Abiotic reductive dechlorination of chlorinated ethylenes by iron bearing soil minerals: 2. Green rust. Environ Sci Technol 36:5147–5154.
Little CD, Palumbo AV, Herbes SE, Lidstrom ME, Tyndall RL, Gilmer PJ. 1988. Trichloroethylene biodegradation by a methane-oxidizing bacterium. Appl Environ Microbiol 54:951–956.
Long C, Borden RC. 2006. Enhanced reductive dechlorination in columns treated with edible oil emulsion. J Contam Hydrol 87:54–72.
Malachowsky KJ, Phelps TJ, Teboli AB, Minnikin DE, White DC. 1994. Aerobic mineralization of trichloroethylene, vinyl chloride, and aromatic compounds by Rhodococcus species. Appl Environ Microbiol 60:542–548.
McDonald JM, Harbaugh AW. 1988. A Modular 3D Finite Difference Ground-Water Flow Model. Technical Report. U.S. Geological Survey Techniques of Water-Resources Investigations, Book 6, Chapter A1, Reston, VA. http://pubs.usgs.gov/twri/twri6a1/html/pdf.html. Accessed June 9, 2009.
McGuire TM, McDade JM, Newell CJ. 2006. Performance of DNAPL source depletion technologies at 59 chlorinated solvent-impact sites. Ground Water Monit Remediat 26:73–84.
Nelson MJK, Montgomery SQ, Pritchard PH. 1988. Trichloroethylene metabolism by microorganisms that degrade aromatic compounds. Appl Environ Microbiol 54:604–606.
Newell CJ, Adamson DT. 2005. Planning-level source decay models to evaluate impact of source depletion on remediation timeframe. Remediat J 15:27–47.
Newell CJ, Rifai HS, Wilson JT, Connor JA, Aziz JJ, Suarez MP. 2002. Calculation and Use of First-Order Rate Constants For Monitored Natural Attenuation Studies. USEPA Remedial Technology Fact Sheet, EPA/540/S-02/500. USEPA National Risk Management Research Laboratory, Cincinnati, OH, USA. November. http://www.epa.gov/ada/pubs/issue.html. Accessed June 9, 2009.
NRC (National Research Council). 2005. Contaminants in the Subsurface: Source Zone Assessment and Remediation. The National Academies Press, Washington, DC, USA. 372 p.
NRC. 2006. Geological and Geotechnical Engineering in the New Millennium: Opportunities for Research and Technological Innovation. National Academies Press, Washington, DC, USA. 222 p.
Parker JC, Park E. 2004. Modeling field-scale dense non-aqueous phase liquid dissolution kinetics in heterogeneous aquifers. Water Resour Res 40:W05109.
Parkhurst DL, Appelo CAJ. 1999. User’s guide to PHREEQC–A computer program for speciation, reaction-path, 1D-transport, and inverse geochemical calculations. Technical Report 99–4259, U.S. Geological Survey Water-Resources Investigations Report. http://wwwbrr.cr.usgs.gov/projects/GWC_coupled/phreeqc/html/final.html. Accessed June 9, 2009.
Peck RB. 1969. Advantages and limitations of the observational method in applied soil mechanics. Geotechnique 19:171–187.
Philp P, Pirkle RJ, McLoughlin PW, Peacock AD, Yang X. 2007. Monitored natural attenuation forum: The use of carbon isotope analysis at MNA sites. Remediat J 17:127–137.
Prommer H. 2002. PHT3D – A Reactive Multicomponent Transport Model for Saturated Porous Media. User’s Manual Version 1.0. Technical Report of Contaminated Land Assessment and Remediation Research Centre. The University of Edinburgh, Edinburgh, UK.
Prommer H, Barry DA, Zheng C. 2003. MODFLOW/MT3DMS based reactive multi-component transport modeling. Ground Water 41:247–257.
Quezada CR, Hansen CM, Clement TP, Jones NL, Lee KK. 2003. ART3D- An analytical model for predicting 3–dimensional reactive transport. Proceedings, MODFLOW and More 2003: Understanding Through Modeling, Golden, Colorado, September 17–19. International Ground Water Modeling Center, Colorado School of Mines, Golden, CO, USA.
Quezada CR, Clement TP, Lee KK. 2004. Generalized solution to multi-dimensional, multi-species transport equations coupled with a first-order reaction network involving distinct retardation factors. Adv Water Resour J 27:507–520.
Rao PSC, Jawitz JW. 2003. Comment on “Steady-state mass transfer from single-component dense non-aqueous phase liquids in uniform flow fields” by T C Sale and D B McWhorter. Water Resour Res 39:1068.
Rao PSC, Annable MD, Sillan RK, Dai D, Hatfield K, Graham WD. 1997. Field-scale evaluation of in situ cosolvent flushing for enhanced aquifer remediation. Water Resour Res 33:2673–2686.
Rao PSC, Jawitz JW, Enfield CG, Falta RW, Annable MD, Wood AL. 2001. Technology integration for contaminated site remediation: Cleanup goals and performance criteria. In Thornton S, Oswald S, eds, Groundwater Quality: Natural and Enhanced Restoration of Groundwater Pollution. Publication No. 275. International Association of Hydrologic Sciences, Wallingford, Oxfordshire, UK, pp 571–578.
Rifai HS, Bedient PB, Wilson JT, Miller KM, Armstrong, JM. 1988. Biodegradation modeling at aviation fuel spill site. J Environ Eng 114:1007–1029.
Saiers JE, Genereux DP, Bolster CH. 2004. Influence of calibration methodology on ground water flow predictions. Ground Water 42:32–44.
Sale TC, McWhorter DB. 2001. Steady state mass transfer from single-component dense non-aqueous phase liquids in uniform flow. Water Resour Res 37:393–404.
Schreiber MD, Feinstein MD, Carey GR, Bahr J. 2004. Physical and chemical mechanisms causing overlap of redox byproducts: Implications for simulating anaerobic biodegradation. J Contam Hydrol 73:99–127.
Schwille F. 1988. Dense Chlorinated Solvents in Porous and Fractured Media: Model Experiments (English Translation). Lewis Publishers, Boca Raton, FL, USA. 175 p.
Steefel CI, MacQuarrie KTB. 1996. Approaches to modeling of reactive transport in porous media. In Lichtner PC, Steefel CI, Oelkers EH, eds, Reactive Transport in Porous Media, Reviews in Mineralogy 34:83–125.
Stroo HF, Unger M, Ward CH, Kavanaugh MC, Vogel C, Leeson A. 2003. Remediating chlorinated solvent source zones. Environ Sci Technol 37:224A–230A.
Suarez MP, Rifai HS. 1999. Biodegradation rates for fuel hydrocarbons and chlorinated solvents in groundwater. Bioremediation J 3:337–362.
Terzaghi K, Peck RN. 1948. Soil Mechanics in Engineering Practice. John Wiley and Sons, New York, NY, USA. 592 p.
Truex MJ, Newell CJ, Looney B, Vangelas K. 2006. Scenarios Evaluation Tool for Chlorinated Solvent MNA. WSRC-STI-2006–00096, Rev. 0. Savannah River National Laboratory, Aiken, SC, USA.
USEPA (U.S. Environmental Protection Agency). 2007. CSMoS Comments on the Potential Limitations of the Domenico-Based Fate and Transport Models, Center for Subsurface Modeling Support (CSMoS), Ada, OK. http://www.epa.gov/ada/csmos/domenico.html. Accessed June 9, 2009.
USEPA. 2008. BIOCHLOR, Version 2.2 Web Site. Center for Subsurface Modeling Support, USEPA National Risk Management Research Laboratory, Ground Water and Ecosystems Restoration Research Division, Ada, OK, USA. http://www.epa.gov/ada/csmos/models/biochlor.html. Accessed June 9, 2009.
Vanderberg LA, Burback BL, Perry JJ. 1995. Biodegradation of trichloroethylene by Mycobacterium vaccae. Can J Microbiol 41:298–301.
Wackett LP, Brusseau GA, Householder SR, Hanson RS. 1989. Survey of microbial oxygenases: Trichloroethylene degradation by propane-oxidation bacteria. Appl Environ Microbiol 55:2960–2964.
Waddill DW, Widdowson MA. 2000. SEAM3D—A numerical model for three dimensional solute transport and sequential electron acceptor-based bioremediation in groundwater. ERDC/EL TR-00–18. U.S. Army Engineer Research and Development Center, Vicksburg, MS, USA.
West MR, Kueper BH, Ungs MJ. 2007. On the use and error of approximation in the Domenico (1987) solution. Ground Water 45:126–135.
Widdowson MA. 2003. SEAM3D: A Numerical Model for Three-Dimensional Solute Transport and Sequential Electron Acceptor-Based Biodegradation in Ground Water. Final Report to the U.S. Army Engineer Research and Development Center, Vicksburg, MS, USA. 110 p.
Widdowson MA, Molz FJ, Benefield LD. 1988. A numerical transport model for oxygen and nitrate-based respiration linked to substrate and nutrient availability in porous media. Water Resour Res 24:1553–1565.
Widdowson MA, Mendez E, Chapelle F, Casey CC. 2004. Natural Attenuation Software, User’s Manual, Version 2. http://www.nas.cee.vt.edu/index.php. Accessed June 9, 2009.
Wiedemeier TH, Swanson MA, Wilson JT, Kampbell DH, Miller RN, Hansen JE. 1996. Approximation of biodegradation rate constants for monoaromatic hydrocarbons (BTEX) in ground water. Ground Water Monit Remediat 16:186–194.
Wiedemeier TH, Rifai HS, Newell CJ, Wilson JT. 1999. Natural Attenuation of Fuels and Chlorinated Solvents in the Subsurface. John Wiley and Sons, New York, NY, USA. 616 p.
Wilson JT, Wilson BH. 1985. Biotransformation of trichloroethylene in soil. Appl Environ Microbiol 49:242–243.
Yeh G-T, Tripathi VS. 1989. A critical evaluation of recent developments in hydrogeochemical transport models of reactive multichemical components. Water Resour Res 25:93–108.
Zawtocki C. 2005. Naturally cleaner groundwater–soybean-based emulsion is proving to decontaminate groundwater more quickly than traditional remediation methods. Mil Eng 97:55–56.
Zhang XH, Bajpai RK. 2000. A comprehensive model for the cometabolism of chlorinated solvents. J Environ Sci Health 35:229–244.
Zhang X, Banerji R, Bajpai R. 1996. Mathematical models for biodegradation of chlorinated solvents: I. Model framework. Proceedings, Hazardous Substance Research Center (HSRC)/Waste-management Education & Research Consortium (WERC) Joint Conference on the Environment, Great Plains/Rocky Mountain HSRC, Manhattan, KS, USA, May 21–23. http://www.engg.ksu.edu/HSRC/96Proceed/. Accessed June 9, 2009.
Zheng C. 1990. MT3DMS. A Modular Three-Dimensional Transport Model for Simulation of Advection, Dispersion and Chemical Reaction of Contaminants in Groundwater Systems. Technical Report. USEPA, Ada, OK, USA. 169 p. http://hydro.geo.ua.edu/mt3d/. Accessed June 9, 2009.
Zheng C, Bennett GD. 1995. Applied Contaminant Transport Modeling: Theory and Practice. Wiley and Sons, Inc., New York, NY, USA. 464 p.
Zheng C, Wang PP. 1999. MT3DMS: A Modular Three-Dimensional Multi-Species Transport Model for Simulation of Advection, Dispersion and Chemical Reactions of Contaminants in Groundwater Systems; Documentation and User’s Guide. U.S. Army Engineer Research and Development Center Contract Report SERDP-99–1, Vicksburg, MS, USA. 202 p.
Zhu J, Sykes JF. 2004. Simple screening models of NAPL dissolution in the subsurface. J Contam Hydrol 72:245–258.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Rifai, H.S., Borden, R.C., Newell, C.J., Bedient, P.B. (2010). Modeling Remediation of Chlorinated Solvent Plumes. In: Stroo, H., Ward, C. (eds) In Situ Remediation of Chlorinated Solvent Plumes. SERDP/ESTCP Environmental Remediation Technology. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-1401-9_6
Download citation
DOI: https://doi.org/10.1007/978-1-4419-1401-9_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-1400-2
Online ISBN: 978-1-4419-1401-9
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)