
Abstract
Activated graphene adsorbents (G-KOH) were synthesized by a one-step alkali-activated method, with a high specific surface area (SSA) and a large number of micropores. As a result, the SSA of the final product greatly increases to ∼512.6 m2/g from ∼138.20 m2/g. The resulting G-KOH was used firstly as an adsorbent for the removal of ciprofloxacin (CIP) in aqueous solutions. Experimental results indicated that G-KOH has excellent adsorption capacity (∼194.6 mg/g). The alkali-activation treatment introduced oxygen-containing functional groups on the surface of G-KOH, which would be beneficial to improving the adsorption affinity of G-KOH for the removal of CIP. Kinetic regression results showed that the adsorption kinetic was more accurately represented by a pseudo-second-order model. The overall adsorption process was jointly controlled by external mass transfer and intra-particle diffusion, and intra-particle diffusion played a dominant role. A Langmuir isotherm model showed a better fit with adsorption data than a Freundlich isotherm model for the adsorption of CIP on G-KOH. The remarkable adsorption capacity of CIP onto G-KOH can be attributed to the multiple adsorption interaction mechanisms (hydrogen bonding, π–π electron donor–acceptor interactions, and electrostatic interactions). Results of this work are of great significance for environmental applications of activated graphene with higher SSA as a promising adsorbent for organic pollutants from aqueous solutions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ciprofloxacin (CIP) as a broad-spectrum synthetic antibiotic widely used for the several bacterial infections, which pose serious threats to the ecosystem and human health due to incomplete metabolism in humans or coming from effluents of drug manufacturers. CIP, even at low concentrations in waters, can lead to the development of antibiotic-resistant bacteria. Even though its removal is of extreme importance, there are not many studies performed on it. Among many chemical, physical, and biological treatment methods, adsorption technology is one of the most effective methods for CIP removal due to its low cost, high efficiency, simplicity, and insensitivity to toxic substances (Gupta and Ali 2008; Gupta et al. 2011, 2008, 2009). A few studies in literature deal with CIP adsorption on activated carbon(Huang et al. 2014), montmorillonite (Wu et al. 2010), carbon nanotubes (CNTs) (Li et al. 2014), and graphene(Wu et al. 2013).
High-capacity adsorbent for the removal of pharmaceutical antibiotics from aqueous solution is still under development to reduce the adsorbent price and to resolve disposal problems. Carbon materials are well-known adsorbents with unique advantages due to their low cost, high adsorption capacity, and easy disposal. CNTs have an ideal limit specific surface area (SSA) of ∼1,300 m2/g. However, the high cost for mass production of high-quality CNTs is a challenge for the commercialization of CNT adsorbents (Ma et al. 2012). Graphene, as a new 2D material with a theoretical SSA of ∼2,630 m2/g, has attracted much attention on the horizon of material science (Ma et al. 2014; Zhu et al. 2011), which has been used recently for gas- and aqueous-phase adsorption as a new and good sorbent candidate. Recently, graphene-based material derived from graphite oxide is now being manufactured in ton quantities at low cost. Peculiarly, the SSA of graphene sheets is related to the number of layers rather than the distribution of pores. However, chemically exfoliated graphene is conventionally prone to intrinsic corrugations and aggregation during the reduction process, consequently only obtaining a relatively low SSA (<100 m2/g).
As adsorbent materials, graphene is required to possess a high SSA and pore volume (PV), which could provide enough adsorption sites for pollutants in wastewater treatment(Ghaedi et al. 2011). To enhance adsorption capacity, physical or chemical activation is required to obtain activated graphene with a higher SSA and more porous structure (Xia et al. 2014; Zhu et al. 2011). In general, physical activation is performed in a CO2 or steam atmosphere at a high temperature, and chemical activation can be achieved using different chemical reagents, typically KOH (Li et al. 2011), H3PO4 (Kuppireddy et al. 2014) or ZnCl2 (Saka 2012). Thus, activation treatment of graphene will provide a good approach to improve the textural properties. However, few investigations have been reported on physical or chemical activation of graphene used as adsorbents.
In this paper, we synthesize an activated graphene adsorbent by a one-step KOH activation annealing method. Obviously, the KOH-activated method can create micropores in the graphene. As a result, the SSA of the final product greatly increases to ∼512.6 m2/g from ∼138.20 m2/g. And then, activated graphene was firstly used as an adsorbent material for removal of selected pharmaceutical antibiotics (CIP) in aqueous solutions. Excellent adsorption properties (∼194.6 mg/g) have been realized. Comparisons of adsorption properties for CIP on graphene before and after activated treatment were further investigated. These results indicate that alkali activation is a useful method to improve the adsorption affinity of CIP contaminants on graphene. Therefore, activated graphene may be a promising adsorbent nanomaterial for organic pollutants from aqueous solutions.
Experimental
Materials
All chemicals were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) in analytical purity and used in the experiments directly without any further purification. All solutions were prepared using deionized water.
Activated treatment of graphene
The graphite oxide was prepared using a modified Hummer’s method (Hirata et al. 2004; Hummers and Offeman 1958; Ma et al. 2014). Graphite oxide was dispersed in deionized water and sonicated in an ultrasound bath for 12 h. The sonicated dispersion was centrifuged for 20 min at 4,000 rpm to remove unexfoliated graphite oxide particles from the supernatant. The obtained suspension of graphene oxide (GO) was then processed by freeze-drying to obtain GO powder. A mixture of GO and KOH powder was performed in a stainless steel vessel in an inert gas atmosphere. The weight ratio of KOH to GO was 4:1. The mixing time was 10 min to obtain a uniform powder mixture. The mixture was then heated to 1,023 K for 1 h under flowing argon in a horizontal tube furnace, washed in concentrated hydrochloric acid and the deionized water, and then dried; the activated GO was noted G-KOH. To compare the performance of adsorbents before and after activated treatment, the GO was also then heated to 1,023 K for 1 h under flowing argon in a horizontal tube furnace, washed in concentrated hydrochloric acid and deionized water, and then dried; the obtained sample was noted G.
Characterization methods
The microstructure and morphology of the G and G-KOH were analyzed by high-resolution transmission electron microscopy (HRTEM, JEOL 2100F, Japan). SSA, PV, and pore size distribution of the G and G-KOH were calculated from the adsorption/desorption isotherms of N2 at 77 K by the multi-point Brunauer–Emmett–Teller (BET), density functional theory (DFT), and Barrett–Joyner–Halenda (BJH) method using an Autosorb-iQ-C (Quantachrome, American, Inc.), and the sample was degassed at 373 K for 4 h prior to the measurements. X-ray photoelectron spectroscopy (XPS) analysis was carried out in a Kratos Axis Ultra DLD spectrometer, using monochromated Al Ka X-rays, at a base pressure of 1 × 10−9 torr. Survey scans determined between 1,100 and 0 eV revealed the overall elemental compositions of the sample, and regional scans for specific elements were performed. The peak energy values were calibrated by placing the major C1s peak at 284.6 eV. Samples were prepared identically to those of the batch experiments. Fourier transform infrared (FT-IR) spectra of powder samples were recorded on a Tensor 27 FT-IR spectrometer (Bruker Optics, Inc.). Raman spectroscopy (Jobin Yvon T64000) was used to further characterize G and G-KOH.
Batch adsorption experiments
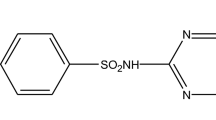
CIP concentration was determined colorimeterically by measuring at maximum absorbance (λ max = 275); the chemical structure of CIP is shown in Fig. S1. A calibration curve was plotted between the absorbance and concentration of the CIP to obtain the absorbance–concentration profile of the CIP based on Beer–Lambert’s law. For high concentration of CIP, it was diluted before absorbance measurement. The concentration of CIP in the solution was determined by the Beer–Lambert’s law expression (Behnajady et al. 2006). Batch adsorption experiments were conducted in 50-ml glass bottles with 20-mg adsorbents and 40-ml CIP solution of different initial concentrations of 40 ∼ 200 mg/L, and the pH of the solution was adjusted to ∼7.0 (nearly in neutral solution to avoid the unpredictable influence) with HCl or NaOH solutions. Timing of the sorption period started as soon as the solution was poured into the bottle. Sample bottles were shaken on a shaker (TS-2102C, Shanghai Tensuclab Instruments Manufacturing Co., Ltd., China) and operated at a constant temperature of 25 °C and at 150 rpm for 3 h. Preliminary experiments indicated that the adsorption of CIP reached equilibrium in ∼1.0 h. Thus, the contact time of 3 h was selected in the batch experiments. All adsorption experiments were conducted in duplicate, and only the mean values were reported. The maximum deviation for the duplicates was usually less than 5 %. The blank experiments without the addition of adsorbents were conducted to ensure that the decrease in the concentration was actually due to the adsorption of adsorbents, rather than by the adsorption on the glass bottle wall. After adsorption, equilibrium has been achieved; the CIP concentrations of the solutions were measured using a spectrophotometer (UV759UV-VIS, Shanghai Precision & Scientific Instrument Co., Ltd.). Kinetic studies were performed at a constant temperature of 25 °C and at 150 rpm with 150 mg/L initial concentration of CIP solutions. The ionic strength experiments were conducted in 150 mg/L CIP solution also at pH 7, with varying concentrations (0.05, 0.1, 0.2, and 0.4 mol/L) of NaCl solution. The effect of solution pH on dye removal was studied in the range of 2–10 with 150 mg/L initial concentrations of CIP solutions. The initial pH values of all the solutions were adjusted using 0.1 mol/L HCl or 0.1 mol/L NaOH solutions with desired concentrations.
The amount of adsorbed dye on adsorbents (q t , mg/g) was calculated as follows:
where c 0 and c t are the dye concentrations at the beginning and after a period of time (mg/L), V is the initial solution volume (L), and m is the adsorbent weight (g).
Results and discussion
Characterization of the adsorbent
The image of GO (Fig. 1a, b) shows that GO is in the form of flexible sheets, which is like silk wave. Figure 1c, d clearly indicates that the activation process etches the graphene and has generated a large destruction of the graphite structure after activated treatment. The large plat of the graphene structure was split into many small plat, which led to the G-KOH agglomeration as shown in Fig. 1c, d.

TEM images in a low and b high magnification of G and c low and d high magnification of G-KOH
The G and G-KOH samples were analyzed by FT-IR for identification of functional groups (Fig. 2a). Some characteristic oxygen groups still remained before and after activation treatment, including C–O stretching (1,113/cm) and C=O and –OH stretching from carbonyl and carboxylic groups (1,631 and 3,435/cm). Activation processes can reduce the GO further and remove the remaining functional oxygen groups partly. These functional groups may also act as anchoring sites for CIP molecules.

FT-IR spectra (a), Raman spectra (b), and XPS survey scans(c) and O1s deconvolution (d) of G and G-KOH
Raman spectroscopy is the most direct and nondestructive technique to characterize the structure and quality of graphene, particularly to determine the defects, the ordered and disordered structures, and the layers of graphene. The Raman spectra for the G and G-KOH samples consist of two peaks at high frequencies (Fig. 2b). The first peak appears at 1,600/cm as the so-called G peak related to E2g graphite mode (Ma and Wang 2008; Ma et al. 2009; Souza Filho et al. 2003). The strong intensity of this peak indicates good graphitization of graphene. The second peak exists at around 1,329/cm as the D peak induced by a defect-related vibration mode (Souza Filho et al. 2003), which is ascribed to disordered carbon, edge defects, and other defects (sp3-bonded carbon, dangling bonds, vacancies, and topological defects). The intensity ratio of the D and G peaks (I D/I G) is an indicator for estimating the quantity of defect in graphene. It clearly shows that there are more defects of G-KOH than those of G from the intensity ratio of the D band and G band (I D/I G = 1.14 and 1.44 for G and G-KOH). The composition of G and G-KOH was determined by XPS as shown in Fig. 2c, d. The G-KOH showed a small O1s signal with an atomic content of 9.14 %, in comparison with 6.61 % for G. This observation reveals minimal introduction of new oxygen-containing functional groups on the surface of G-KOH compared with G.
The N2 adsorption/desorption isotherms and pore distribution of G and G-KOH are presented in Fig. 3. In comparison, the N2 adsorption/desorption amount of G-KOH is considerably higher than that of G at low or high pressure. The detailed features of mesopore and micropore analyzed by the BJH and DFT method are presented in Table 1. The SSA of G-KOH drastically increased by about ∼3.7 times than G. Such increases correspond to a decrease in the mean pore diameter from ∼3.946 to ∼1.658 nm. Moreover, the micro-PV of KOH is improved almost by ∼3.7 times than G.

N2 adsorption/desorption isotherms (a) and pore size distribution (b) of G and G-KOH
The abovementioned results indicated that alkali activation is a high-efficiency technique for producing a large scale of porous structures. The activation mechanism is normally suggested to include independent hydroxide and redox processes during the reaction. With the activation treatment, KOH powder can react with carbon as follows (Lillo-Rodenas et al. 2003, 2004; Ma et al. 2012):
When the temperature is higher than 700 °C, the reaction proceeds as follows:
When the temperature is higher than 800 °C, the reaction proceeds as follows:
The aggregation of graphene will also reduce the useful SSA. After KOH-activated treatment, the flat of graphene was destroyed and decreased, and the dispersion is strongly improved. Most importantly, the graphitic structure of graphene will be destroyed. Large quantities of new micropores are produced. This implies that the graphene after the KOH-activation treatment possesses more micropores and could give rise to an obvious improvement in total SSA.
Effect of dosage
The experimental results of dosage effect for CIP on G-KOH are shown in Fig. 4. It was observed that the adsorption capacity of CIP decreased as the G-KOH dosage was increased over the range 100 to 400 mg/L. High G-KOH dosage may increase the viscosity and inhibit the diffusion of CIP molecules to the surface of G-KOH. Considering the cost, operability, adsorption capacity, and accurate weigh, the adsorbent dosage was selected as 20 mg.

Effect of adsorbent dosage on CIP adsorption: initial concentration 150 mg/L, contact time 24 h, and agitation speed 150 r/min
Adsorption isotherms
Figure 5 shows the equilibrium isotherms for adsorption of CIP onto G and G-KOH, and the equilibrium adsorption characteristics were analyzed by using Langmuir (Langmuir 1918) and Freundlich (Freundlich 1906) isotherm models. The isotherms based on the experimental data and the parameters obtained from nonlinear regression by both models are shown in Fig. 5. The determination coefficients (R 2) of the Langmuir and Freundlich isotherms of G and G-KOH are summarized in Table 2. The Freundlich isotherm model showed a better fit with adsorption data than the Langmuir isotherm model for G based on the determination coefficient. The applicability of the Freundlich isotherm suggests that different sites with several adsorption energy values are involved, and in some cases, the intermolecular interactions occur between CIP and G. However, the result is in inverse for K-KOH.

Equilibrium adsorption isotherms of CIP on G and G-KOH
Figure 5 shows that the computed maximum monolayer capacities are 145.9 mg/g for CIP on G and 194.6 mg/g for CIP on G-KOH. The adsorption capacity of CIP increased obviously almost by ∼1.33 times after KOH-activated treatment. The above results might be caused by the significant differences of SSA between G (138.20 m2/g) and G-KOH (512.65 m2/g). From the composition analysis of adsorbents, the oxygen atomic content of G-KOH increased from 6.61 to 9.14 % after the activated treatment (as shown in Fig. 2b). This result indicates that the surface of G-KOH was modified by more oxygen-containing functional groups. Here, the surface oxygen content may affect the dispersibility of G-KOH and the water cluster formation in aqueous solution, which consequently can be favorable for the aqueous phase adsorption (Yu et al. 2012). With the increasing of oxygen content, maximum adsorption capacity is significantly enhanced. Similar results have been reported (Yu et al. 2011). Because CIP has good solubility, the main purpose of the surface functionalization of graphene is to improve its hydrophilicity and dispersibility in aqueous solutions. A better dispersion of the G-KOH in water will increase the available adsorption sites, which may be favorable for the CIP adsorption in aqueous solution.
The R L value of G is 0.027 for CIP; the R L value of G-KOH is 0.057 for CIP, indicating that the adsorption of CIP onto G and G-KOH is favorable, and the G and G-KOH are an excellent adsorbent material for CIP removal from aqueous solution. To deepen the understanding of adsorption mechanism, Dubinin–Radushkevich (D-R) isotherm model was chosen to describe adsorption on both homogenous and heterogeneous surfaces (Jovanovic et al. 2011). The D-R isotherm model was also applied to distinguish between physical and chemical adsorption of CIP on G and G-KOH. The isotherm constant, E, and determination coefficients are calculated and presented in Table 2. The values of E are below 5 kJ/mol for the entire range of investigated CIP concentration. The value of this parameter can give important information about adsorption mechanism (Onyango et al. 2004; Tahir and Rauf 2006). The mean energy of CIP adsorption on G and G-KOH is below 5 kJ/mol calculated by D-R isotherm model, which suggests that physical adsorption is dominating in the adsorption process between the CIP and adsorbents.
Adsorption kinetics
To study the adsorption kinetics of CIP on G and G-KOH, 150 mg/L initial concentrations of corresponding CIP solutions were used. The adsorption removal of CIP on G-KOH was found to be rapid at the initial period (∼10 min) and then became slow and stagnate with the increase in contact time (∼10 to ∼80 min) and nearly reached a plateau after approximately 80 min of the experiment as shown in Fig. 6a. Generally, the removal rate of pollutants is rapid initially, but it gradually decreases with time until it reaches equilibrium. This phenomenon is attributed to the fact that a large number of vacant adsorption sites are available for adsorption at the initial stage, and after a lapse of time, the remaining vacant adsorption sites are difficult to be occupied due to repulsive forces between the solute molecules on the solid and bulk phases (Apul et al. 2012; Ghaedi et al. 2011). Contact time is one of the most important parameters for practical applications. It was found that the G-KOH and G showed a much higher adsorption rate than that of a commercial activated carbon (Iriarte-Velasco et al. 2011) and other adsorbents (Crini 2006).

Kinetic curves (a), pseudo-second-order model (b), Weber–Morris model (c), and Boyd model (d) of G and G-KOH
Adsorption is a physicochemical process that involves mass transfer of a solute from liquid phase to the adsorbent surface. In order to understand the characteristics of the adsorption process, the pseudo-first-order (PFO) and pseudo-second-order (PSO) kinetic models were applied to fit experimental data obtained from batch experiments, as shown in Figs. 6 and S2. The kinetic parameters and the determination coefficients (R 2) were determined by nonlinear regression and are given in Table 3. The R 2 values of the PSO kinetic model are much higher than those of PFO, the calculated q e values (q e,cal ) of PSO models are close to the experimental ones (q e,exp ). Hence, the PSO kinetic model is more appropriate to describe the adsorption behavior of CIP onto G and G-KOH.
If the movement of adsorbate from the bulk liquid to the liquid film surrounding the adsorbent is ignored, the adsorption process in porous solids can be separated into three stages as follows (Ma et al. 2012; Rengaraj et al. 2004): (1) external mass transfer of the adsorbate across the liquid film to the adsorbent exterior surface, which is also called film diffusion (or boundary layer diffusion or outer diffusion), (2) transport of the adsorbate from the adsorbent exterior surface to the pores or capillaries of the adsorbent internal structure, which is called intra-particle diffusion (or inner diffusion), and (3) the adsorbate is adsorbed onto the active sites in inner and outer surfaces of the adsorbent. The third step is considered to be very fast and thus cannot be treated as rate-limiting step. Generally, the adsorption rate is controlled by outer or inner diffusion or both. In order to determine the actual rate-controlling step involved in the CIP sorption process, a Weber–Morris equation was applied. Plots of q t against t 1/2 are shown in Fig. 6c, and the corresponding kinetic parameters are listed in Table 3. It was also observed that the regression of q t versus t 1/2 was not linear, and the plots do not pass through the origin, suggesting that the intra-particle diffusion is not the sole rate-controlling step (Dogan et al. 2009), and the external mass transfer may also be significant in the rate-controlling step due to the large intercepts of the linear portion of the plots (Al-Degs et al. 2006).
The adsorption kinetic data were further analyzed by a Boyd model (Kumar et al. 2005). The calculated B t values were plotted against time t as shown in Fig. 6d. The linearity of the plots provides useful information to distinguish between external mass transfer and intra-particle-diffusion-controlled mechanism of adsorption (Sankar et al. 1999). The plots in Fig. 6d do not pass through the origin, confirming the involvement of external mass transfer in the entire adsorption process (Gupta et al. 2008). However, the plots are not straight lines, indicating that external mass transfer shows a relatively weak rate control for CIP adsorption onto G-KOH. So, the overall adsorption process may be jointly controlled by external mass transfer and intra-particle diffusion.
Effect of pH and ionic strength
The adsorption process possibly involved in many mechanisms included electrostatic interactions, electron donor–acceptor (EDA) interaction, hydrogen bond, etc. Figure 7a shows the adsorption capacity of CIP on G-KOH at different ionic strengths (0–0.4 M). As the ionic strength was increased from 0 to 0.2 M, the adsorption capacity of CIP adsorbed increased from 187 to 241 mg/g. With the increase of ionic strength, the adsorption capacity of CIP adsorbed has remained unchanged; there may be existing cooperative adsorption behavior between CIP and NaCl.

Effect of ionic strength (a) and pH values (b) on adsorption capacity of CIP on G-KOH (CIP concentration = 150 mg/L, G-KOH = 0.5 g/L)
Generally, adsorption of ionizable organic contaminants is affected by pH due to the varied species. CIP has positively charged (cationic), negatively charged (anionic), and/or zwitterionic species at different pH due to different pKa (6.1 and 8.7). The cationic CIP was dominant at pH <6.1, while the dominant species was anion at pH >8.7. In this study, the adsorption capacity decreased at pH <6 or pH >8 and remained unchanged between pH 6–8, which is consist with the ionic station of CIP, indicating that the electrostatic interaction may be a controllable mechanism. The main differences between G and G-OH were their SSA and functional groups. The functional group was one factor responsible for CIP sorption on graphene. It was observed that oxygen functional groups on graphene surface including –OH and –COOH groups could reduce the hydrophobicity of graphene surface. When the pH is ∼7, the zwitterionic species mainly exist, so the oxygen-containing functional groups have little effect on the electrostatic interaction between CIP and G-KOH, and the physical adsorption mechanism based on the SSA may play a critical role in the process of adsorption; the results are also consistent with the D-R analysis and SSA results.
The adsorption mechanism of CIP on G-KOH can also be explained by π–π EDA interaction, one of the driving forces for the sorption of organic chemicals with benzene rings on graphene. The benzene rings with fluorine group and N-heteroaromatic ring in CIP can function as a π electron acceptor due to the strong electron-withdrawing ability of N and F, as shown in Fig. S1. The –OH groups on the G-KOH surface can make the benzene rings of G-KOH act as electron donors. Thus, significantly enhanced CIP sorption on G-KOH by formation of a π–π bond was expected. However, the π–π bond was significantly depressed between CIP and G-KOH because the –COOH groups make G-KOH electron acceptors. Hydrogen bond widely exists in the process of polar organic pollutant sorption on carbon nanomaterials (Yang et al. 2008). Functional groups of organic chemicals can act as hydrogen-bonding donors and form hydrogen bonds with the graphite sheets of graphene, where the benzene rings can act as hydrogen-bonding acceptors. CIP hold C=O and –OH groups as hydrogen-bonding donors which can form hydrogen bonds with the oxygen-containing functional groups on graphene surface (Wang et al. 2010).
Conclusion
In this study, we synthesize an activated graphene adsorbent by a one-step KOH activation method. Obviously, the SSA of the final product greatly increases to ∼512.6 m2/g from ∼138.20 m2/g. And then, activated graphene was firstly used as an adsorbent for the removal of CIP in aqueous solutions. The maximum adsorption capacity (∼194.6 mg/g) has been realized, which indicates that alkali activation is a useful method to improve the adsorption affinity of CIP contaminants on graphene. Therefore, activated graphene may be a promising adsorbent nanomaterial for organic pollutants from aqueous solutions.
References
Al-Degs YS, El-Barghouthi MI, Issa AA, Khraisheh MA, Walker GM (2006) Sorption of Zn(II), Pb(II), and Co(II) using natural sorbents: equilibrium and kinetic studies. Water Res 40:2645–2658
Apul OG, Shao T, Zhang SJ, Karanfil T (2012) Impact of carbon nanotube morphology on phenanthrene adsorption. Environ Toxicol Chem 31:73–78
Behnajady MA, Modirshahla N, Fathi H (2006) Kinetics of decolorization of an azo dye in UV alone and UV/H2O2 processes. J Hazard Mater 136:816–821
Crini G (2006) Non-conventional low-cost adsorbents for dye removal: a review. Bioresour Technol 97:1061–1085
Dogan M, Abak H, Alkan M (2009) Adsorption of methylene blue onto hazelnut shell: kinetics, mechanism and activation parameters. J Hazard Mater 164:172–181
Freundlich HMF (1906) Over the adsorption in solution. J Phys Chem 57:385–471
Ghaedi M, Hassanzadeh A, Kokhdan SN (2011) Multiwalled carbon nanotubes as adsorbents for the kinetic and equilibrium study of the removal of Alizarin Red S and Morin. J Chem Eng Data 56:2511–2520
Gupta VK, Ali I (2008) Removal of endosulfan and methoxychlor from water on carbon slurry. Environ Sci Technol 42:766–770
Gupta VK, Mittal A, Gajbe V, Mittal J (2008) Adsorption of basic fuchsin using waste materials-bottom ash and deoiled soya-as adsorbents. J Colloid Interface Sci 319:30–39
Gupta VK, Mittal A, Malviya A, Mittal J (2009) Adsorption of carmoisine A from wastewater using waste materials-Bottom ash and deoiled soya. J Colloid Interface Sci 335:24–33
Gupta VK, Gupta B, Rastogi A, Agarwal S, Nayak A (2011) A comparative investigation on adsorption performances of mesoporous activated carbon prepared from waste rubber tire and activated carbon for a hazardous azo dye-Acid Blue 113. J Hazard Mater 186:891–901
Hirata M, Gotou T, Horiuchi S, Fujiwara M, Ohba M (2004) Thin-film particles of graphite oxide 1: high-yield synthesis and flexibility of the particles. Carbon 42:2929–2937
Huang LH, Wang M, Shi CX, Huang J, Zhang B (2014) Adsorption of tetracycline and ciprofloxacin on activated carbon prepared from lignin with H3PO4 activation. Desalin Water Treat 52:2678–2687
Hummers WS Jr, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339
Iriarte-Velasco U, Chimeno-Alanis N, Gonzalez-Marcos MP, Alvarez-Uriarte JI (2011) Relationship between thermodynamic data and adsorption/desorption performance of acid and basic dyes onto activated carbons. J Chem Eng Data 56:2100–2109
Jovanovic BM, Vukasinovic-Pesic VL, Rajakovic LV (2011) Enhanced arsenic sorption by hydrated iron (III) oxide-coated materials-mechanism and performances. Water Environ Res 83:498–506
Kumar KV, Ramamurthi V, Sivanesan S (2005) Modeling the mechanism involved during the sorption of methylene blue onto fly ash. J Colloid Interface Sci 284:14–21
Kuppireddy SKR, Rashid K, Al Shoaibi A, Srinivasakannan C (2014) Production and characterization of porous carbon from date palm seeds by chemical activation with H3PO4: process optimization for maximizing adsorption of methylene blue. Chem Eng Commun 201:1021–1040
Langmuir I (1918) The adsorption of gases on plane surfaces of glass, mica and platinum. J Am Chem Soc 40:1361–1403
Li YM, van Zijll M, Chiang S, Pan N (2011) KOH modified graphene nanosheets for supercapacitor electrodes. J Power Sources 196:6003–6006
Li HB, Zhang D, Han XZ, Xing BS (2014) Adsorption of antibiotic ciprofloxacin on carbon nanotubes: pH dependence and thermodynamics. Chemosphere 95:150–155
Lillo-Rodenas MA, Cazorla-Amoros D, Linares-Solano A (2003) Understanding chemical reactions between carbons and NaOH and KOH - an insight into the chemical activation mechanism. Carbon 41:267–275
Lillo-Rodenas MA, Juan-Juan J, Cazorla-Amoros D, Linares-Solano A (2004) About reactions occurring during chemical activation with hydroxides. Carbon 42:1371–1375
Ma J, Wang JN (2008) Purification of single-walled carbon nanotubes by a highly efficient and nondestructive approach. Chem Mater 20:2895–2902
Ma J, Wang JN, Wang XX (2009) Large-diameter and water-dispersible single-walled carbon nanotubes: synthesis, characterization and applications. J Mater Chem 19:3033–3041
Ma J, Yu F, Zhou L, Jin L, Yang MX, Luan JS, Tang YH, Fan HB, Yuan ZW, Chen JH (2012) Enhanced adsorptive removal of methyl orange and methylene blue from aqueous solution by alkali-activated multiwalled carbon nanotubes. ACS Appl Mater Inter 4:5749–5760
Ma J, Zhou L, Li C, Yang JH, Meng T, Zhou HM, Yang MX, Yu F, Chen JH (2014) Surfactant-free synthesis of graphene-functionalized carbon nanotube film as a catalytic counter electrode in dye-sensitized solar cells. J Power Sources 247:999–1004
Onyango MS, Kojima Y, Aoyi O, Bernardo EC, Matsuda H (2004) Adsorption equilibrium modeling and solution chemistry dependence of fluoride removal from water by trivalent-cation-exchanged zeolite F-9. J Colloid Interface Sci 279:341–350
Rengaraj S, Kim Y, Joo CK, Yi J (2004) Removal of copper from aqueous solution by aminated and protonated mesoporous aluminas: kinetics and equilibrium. J Colloid Interface Sci 273:14–21
Saka C (2012) BET, TG-DTG, FT-IR, SEM, iodine number analysis and preparation of activated carbon from acorn shell by chemical activation with ZnCl2. J Anal Appl Pyrol 95:21–24
Sankar M, Sekaran G, Sadulla S, Ramasami T (1999) Removal of diazo and triphenylmethane dyes from aqueous solutions through an adsorption process. J Chem Technol Biotechnol 74:337–344
Souza Filho A, Jorio A, Samsonidze GG, Dresselhaus G, Pimenta M, Dresselhaus M, Swan AK, Ünlü M, Goldberg B, Saito R (2003) Competing spring constant versus double resonance effects on the properties of dispersive modes in isolated single-wall carbon nanotubes. Phys Rev B 67:035427
Tahir SS, Rauf N (2006) Removal of a cationic dye from aqueous solutions by adsorption onto bentonite clay. Chemosphere 63:1842–1848
Wang ZY, Yu XD, Pan B, Xing BS (2010) Norfloxacin sorption and its thermodynamics on surface-modified carbon nanotubes. Environ Sci Technol 44:978–984
Wu QF, Li ZH, Hong HL, Yin K, Tie LY (2010) Adsorption and intercalation of ciprofloxacin on montmorillonite. Appl Clay Sci 50:204–211
Wu SL, Zhao XD, Li YH, Zhao CT, Du QJ, Sun JK, Wang YH, Peng XJ, Xia YZ, Wang ZH, Xia LH (2013) Adsorption of ciprofloxacin onto biocomposite fibers of graphene oxide/calcium alginate. Chem Eng J 230:389–395
Xia BY, Yan Y, Wang X, Lou XW (2014) Recent progress on graphene-based hybrid electrocatalysts. Mater Horiz 1:379–399
Yang K, Wu WH, Jing QF, Zhu LZ (2008) Aqueous adsorption of aniline, phenol, and their substitutes by multi-walled carbon manotubes. Environ Sci Technol 42:7931–7936
Yu F, Ma J, Wu YQ (2011) Adsorption of toluene, ethylbenzene and m-xylene on multi-walled carbon nanotubes with different oxygen contents from aqueous solutions. J Hazard Mater 192:1370–1379
Yu F, Wu YQ, Ma J (2012) Influence of the pore structure and surface chemistry on adsorption of ethylbenzene and xylene isomers by KOH-activated multi-walled carbon nanotubes. J Hazard Mater 237:102–109
Zhu YW, Murali S, Stoller MD, Ganesh KJ, Cai WW, Ferreira PJ, Pirkle A, Wallace RM, Cychosz KA, Thommes M, Su D, Stach EA, Ruoff RS (2011) Carbon-based supercapacitors produced by activation of graphene. Science 332:1537–1541
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 21207100 and No. 51408362), Specialized Research Fund for the Doctoral Program of Higher Education (20120072120058), the Shanghai Institute of Technology Scientific Research Foundation for Introduced Talent, China (No. YJ2014-30), the Key Research Projects of the Science and Technology Commission of Shanghai Municipality (12231201702), and the Central Finance Special Fund to Support the Development of Local Colleges and Universities (City Safety Engineering). We are also thankful to anonymous reviewers for their valuable comments to improve this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editor: Philippe Garrigues
Fei Yu and Jie Ma contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 116 kb)
Rights and permissions
About this article

Cite this article
Yu, F., Ma, J. & Bi, D. Enhanced adsorptive removal of selected pharmaceutical antibiotics from aqueous solution by activated graphene. Environ Sci Pollut Res 22, 4715–4724 (2015). https://doi.org/10.1007/s11356-014-3723-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-014-3723-9