
Abstract
Purpose
Obstructive sleep apnea hypopnea syndrome has been reported to be associated with pulmonary hypertension (PH). Adiponectin (Ad) has many protective roles in the human body, including its function as an anti-inflammatory and an anti-oxidant, as well as its role in preventing insulin resistance and atherosclerosis. This study aimed to investigate the molecular mechanism of chronic intermittent hypoxia (CIH)-induced pulmonary injury and the protective role of Ad in experimental rats.
Methods
Thirty male Sprague-Dawley rats were randomly divided into three groups with 10 rats in each group: normal control (NC) group, CIH group, and CIH + Ad group. Rats in the NC group were kept breathing room air for 12 weeks. Rats in the CIH group were intermittently exposed to a hypoxic environment for 8 h/day for 12 weeks. Rats in the CIH + Ad group received 10 μg Ad twice weekly via intravenous injection. After 12 weeks of CIH exposure, we detected the pulmonary function, pulmonary artery pressure, lung histology, pulmonary cell apoptosis, pulmonary artery endothelial cell apoptosis, mitochondrial membrane potential (MMP), and reactive oxygen species (ROS) level. We also analyzed expression proteins involved in the mitochondria-, endoplasmic reticulum (ER) stress-, and Fas receptor-associated pulmonary apoptosis pathways, as well as the SIRT3/SOD2 pathway.
Results
CIH exposure for 12 weeks did not lead to abnormal pulmonary function, PH, or pulmonary artery endothelial cell apoptosis. However, we observed a significant increase in the rate of pulmonary cell apoptosis, the expression of proteins involved in mitochondria-, ER stress-, and Fas receptor-associated pulmonary apoptosis pathways, and the generation of ROS in the CIH group compared with the NC group. In contrast, the MMP and protein expressions of SIRT3/SOD2 pathway were significantly decreased in the CIH group compared with the NC group. Ad supplementation in the CIH + Ad group partially improved these changes induced by CIH.
Conclusion
Even though CIH did not cause abnormal pulmonary function or PH, early lung injury was detected at the molecular level in rats exposed to CIH. Treatment with Ad ameliorated the pulmonary injury by activating the SIRT3/SOD2 pathway, reducing ROS generation, and inhibiting ROS-associated lung cell apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea hypopnea syndrome (OSAHS) is a common clinical condition characterized by recurrent episodes of partial or complete upper airway obstructions during sleep, which promotes intermittent hypoxia and sleep fragmentation [1]. It is well-known that OSAHS increases the risk of hypertension and diabetes [2] and is also believed to have a significant hemodynamic impact [3], which may play a role in the development of pulmonary hypertension (PH) [4]. Chronic intermittent hypoxia (CIH), a major component of OSAHS, may play the most important role in the pathogenic onset of PH in OSAHS patients. Long-term CIH leads to pulmonary vessel systole, endothelial dysfunction, pulmonary vascular reconstruction, and ultimately, PH [4, 5]. Fagan et al. found that CIH induced pulmonary vascular remodeling and PH in mice [6], and Hamada et al. reported that CIH induced mitochondrial hydrogen peroxide in cultured airway epithelial cells [7]. We hypothesized that CIH might induce pulmonary injury, but the potential mechanism requires further investigation. The production of reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals is normally kept in balance but is increased under conditions of hypoxia or starvation, and an excess of ROS has been associated with a variety of illnesses [8, 9]. Increased ROS production after CIH [10] may further contribute to pulmonary injury.
Adiponectin (Ad), as an adipocytokine, is abundant in serum. In the plasma, Ad exists as dimers, trimers, or hexamers; Ad is also found in plasma as full-length or globular adiponectin. All forms of Ad have bioactivity [11]. In the human body, Ad functions as both an anti-inflammatory and an anti-oxidant and also plays an important role in the prevention of insulin resistance and atherosclerosis [12, 13]. It has been reported that serum Ad levels are decreased in OSAHS patients and are negatively related to the apnea hypoxia [14]. Thus, we speculated that the decreased Ad levels observed in OSAHS patients might contribute to CIH-induced pulmonary injury. Kim found that globular adiponectin maintained epidermal homeostasis by affecting epidermal melanocyte biology and protected hepatic cells against acetaminophen-induced apoptosis [15, 16]. In this study, we focused on the lung protective effect of globular adiponectin. The present study aimed to investigate changes in pulmonary function and pulmonary artery pressure, as well as the molecular pulmonary damage induced by CIH. Furthermore, we aimed to elucidate the protective role of Ad and its related mechanism.
Material and methods
Animals and CIH exposure
A total of 30 male Sprague-Dawley rats (specific pathogen free, Shanghai Solake Ltd) aged 8 weeks were housed in the Animal Care Center of Southeast University. The study was approved by the Animal Ethics Committee of Southeast University. All experimental protocols and relevant details were approved by the Animal Ethics Committee of Southeast University.
The CIH rat model was used to study the pathophysiology of OSAHS and has been described in detail in our previous reports [17, 18]. The rats were randomly allocated to normal control (NC), CIH, and CIH + Ad groups. Each group had 10 rats. Rats in the CIH and CIH + Ad groups were housed in cages in which the fraction of inspired oxygen (FiO2) was gradually decreased to 4–5% for 15 s and then gradually increased to 21% for 15 s. The length of each cycle was 2 min. The CIH exposure cycle was repeated 30 times per hour and continued for 8 h per day to mimic moderate OSAHS. Rats in the NC group were housed in cages with normal atmospheric oxygen concentration (21%). Rats in the CIH + Ad group received tail vein injections of murine Ad (Biovision, Inc. USA. Catalog #: 4593) injection at a dose of 10 μg, twice per week. The Ad administered was recombinant globular adiponectin, with a molecular weight of 16.6 kDa. The Ad was endotoxin-free and was diluted to 20 μg/mL in saline. The rats in the NC and CIH groups received tail vein injections of 0.5 mL of saline, twice per week. These experimental procedures were carried out for 12 weeks.
Pulmonary function and pulmonary artery pressure
After pentobarbital anesthesia and endotracheal intubation, rats were placed in a closed chamber and ventilated with an animal respirator. The pressure changes within the chamber were converted to an electrical signal and transferred to the respirator. The inhalation resistance (Ri), exhalation resistance (Re), pulmonary compliance (CL), and 0.3-s forced expiratory volume/forced vital capacity (FEV0.3/FVC) were measured by the respirator.
After pulmonary function testing, pulmonary artery pressure was measured. A midline neck incision was made, the right external jugular vein and left common carotid artery were dissected, and the catheter was inserted into the pulmonary artery and right ventricle via the right external jugular vein. The catheter was connected to a biological data analysis system that recorded pressure changes in the right ventricle and pulmonary artery. The average pulmonary artery pressure was calculated.
Hematoxylin and eosin staining
The rats were sacrificed by decapitation after measuring the pulmonary artery pressure. Pulmonary tissue samples were obtained and fixed in 4% paraformaldehyde for 48 h. The tissues were dehydrated, paraffin embedded, and sectioned at 5 μm for HE staining. Briefly, the sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin for 1 min each. After dehydration and cover slipping, the stained tissue sections were observed by light microscopy. In this study, each lung sample was evaluated in five high power fields (200×) independently by two investigators in blinded fashion per lung sample.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Transferase dUTP nick end labeling (TUNEL) staining was performed following the kit manufacturer’s (Roche, Germany) instructions. After deparaffinization and rehydration, the pulmonary tissue sections were infused with protein K (20 μg/mL) for 15 min at room temperature and stained with the TUNEL mixture for 60 min at 37 °C in a dark humidified atmosphere, followed by TUNEL-POD staining for 30 min at 37 °C in a dark humidified atmosphere. After staining with diaminobenzidine and hematoxylin for 1 min, cover slips were mounted to the sections. TUNEL-positive cells were counted by light microscopy. Any brown cell nucleus was counted as TUNEL-positive whereas any purple cell nucleus was counted as a normal nucleus. Three fields of vision were chosen; purple and brown nuclei were counted separately. The ratio of TUNEL-positive cells/normal cells was calculated.
Measurement of mitochondrial membrane potential
The JC-1 kit (GENMED, China) was used to detect the pulmonary mitochondrial membrane potential (MMP) in the three groups. Briefly, fresh pulmonary tissue was embedded in OCT and cut into 8-μm sections. A working solution was prepared by adding 10 μL reagent B into 1 mL reagent C. The sections washed with reagent A and immersed into preheated working solution in a dark environment at 37 °C for 20 min. The sections were again washed with reagent A and then mounted with cover slips. The lung sections were visualized using fluorescence microscopy (Olympus, Tokyo, Japan). Image J was used to evaluate red and green fluorescence intensity of three fields per lung sample.
Dihydroethidium staining
Dihydroethidium (DHE) (Beyotime Biotech, China) was used to assess ROS levels in lung tissue. Fresh pulmonary tissue was embedded in OCT, frozen, and cut into 8-μm sections. The lung tissue sections were immersed in DHE (10 μg/mL) at 37 °C for 15 min in a humidified environment. The lung tissue sections were washed three times with PBS and visualized using fluorescence microscopy (Olympus, Tokyo, Japan). Image J was used to evaluate the red fluorescence intensity of three fields per lung sample.
Western blot assay
Total protein was extracted from lung tissues using a Tissue Protein Extraction Reagent kit (Thermo Fisher Scientific, Rockford, IL, USA) containing 10× phosphatase inhibitor cocktail (Roche, Germany) and 1 mM phenylmethylsulfonyl fluoride.
Cytosolic and mitochondrial fractions were obtained from lung cells using a mitochondria extraction kit (Beyotime, China) according to the manufacturer’s instructions. The tissues (50 mg) were cut into small pieces after washing with PBS and infused into 500 μL mitochondrial isolation reagent A containing 1 mM PMSF. A dounce homogenizer was used to homogenize the lung tissue on ice. The lung suspensions were centrifuged at 600g for 5 min at 4 °C, and the supernatant was collected. The supernatant was then centrifuged at 3500g for 10 min at 4 °C. The supernatant (cytosolic fraction) and the mitochondrial pellet were collected separately. Then, the mitochondria were lysed in 2% CHAPS in Tris-buffered saline (TBS; 25 mM Tris, 0.15 M NaCl; pH 7.2). The supernatant then was centrifuged at 12,000g for 10 min at 4 °C and the supernatant (cytosolic fraction) was collected. The cytosolic fractions were used to quantify cytosolic cytochrome c; the mitochondrial fraction was used to detect mitochondrial cytochrome c.
The protein concentration was determined using a bicinchoninic acid assay kit (Thermo Fisher Scientific, USA). Equal quantities of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. After blocking with 5% bovine serum albumin for 1 h, the membranes were incubated overnight with primary antibodies at 4 °C and then with secondary antibodies for 1 h at 37 °C. The protein blots were visualized using an enhanced chemiluminescence kit (Thermo Fisher Scientific, San Jose, CA, USA) and were read using a digital imaging system (ChemiDoc XRS+ System; Bio Rad, Hercules, CA, USA). Primary antibodies against Bax (#2772), bcl-2 (#2876), cytochrome C (#4272), GRP78 (#3183), TRAF2 (#4712), p-JNK (#9255), JNK (#9252), p-P38 MAPK (#9216), P38 MAPK (#9212), CHOP (#2895), SIRT3 (#5490), and SOD2 (#13194) were purchased from Cell Signaling Technology (CST; Danvers, MA, USA). Primary antibodies against IRE1 (ab37073), p-IRE1 (ab48187), caspase-12 (ab62463), FAS (ab82419), FASL (ab15285), FADD (ab24533), and Acetylated-SOD2 (Ac-SOD2; ab137037) were purchased from Abcam (Burlingame, CA, USA). The primary antibody against β-actin (sc70319) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Secondary antibodies (#7074 and #7076) were purchased from CST.
Statistical analysis
All results are reported as mean ± standard error of the mean of three independent assays. Significant differences among the three study groups were determined by one-way analysis of variance. Post hoc analysis was performed using the Student–Newman–Keuls test. A p value < 0.05 was considered significant. All analyses were two-tailed and performed using SPSS version 19.0 (SPSS, Inc. Chicago IL).
Results
Pulmonary function and PH
The pulmonary function and PH were detected in the three groups 12 weeks. As shown in Table 1, at the end of the experiment, there were no differences in Ri, Re, CL, Fev0.3/FVC, PASP, PADP, or mPAP among the three groups (Table 1; all p values > 0.05).
Pulmonary histology, pulmonary cell apoptosis, and pulmonary artery endothelial cell apoptosis
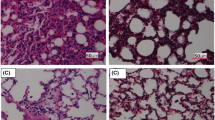
The pulmonary histological tissues of the three groups were evaluated in HE-staining (original magnification × 200). As shown in Fig. 1a, when compared to the NC group, CIH exposure induced interstitial pulmonary edema and infiltration of inflammatory cells in alveoli. Supplementation with Ad partly reduced the interstitial pulmonary edema and infiltration of inflammatory cells in alveoli compared to the CIH group. As shown in Fig. 1c, there was no difference in pulmonary artery histology among the three experimental groups.

Histological changes. Supplementation with Ad reduced CIH-induced interstitial pulmonary edema and infiltration of inflammatory cells in alveoli and pulmonary cell apoptosis. There was no difference among three groups with respect to pulmonary artery endothelial cell apoptosis. a Morphology of lung tissue in the three study groups as shown by HE staining (original magnification × 200). b Pulmonary cell apoptosis assayed by TUNEL staining (original magnification × 400). c Pulmonary artery morphology in the three study groups as shown by HE staining (original magnification × 200). d Pulmonary artery endothelial cell apoptosis assayed by TUNEL staining (original magnification × 400). NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars show ± SD. *p < 0.05
TUNEL staining (original magnification × 400) was used to assess rates of apoptosis rates in pulmonary cells and endothelial cells across the three groups. As shown in Fig. 1b, the pulmonary cell apoptosis rate was higher in the CIH group (6.962 ± 0.7766%) than in the NC group (0.796 ± 0.0594%; p < 0.05). The pulmonary cell apoptosis rate in the CIH + Ad group (2.552 ± 0.8274%) was lower than in the CIH group (6.962 ± 0.7766%; p < 0.05), but higher than that it in the NC group (0.796 ± 0.0594%; p < 0.05). As shown in Fig. 1d, there were no differences among the three groups in the apoptosis rate of pulmonary artery endothelial cells (NC vs CIH vs CIH + Ad: 0.915 ± 0.021% vs 0.930 ± 0.054% vs 0.942 ± 0.102%; p < 0.05).
MMP level and ROS generation
The MMP level was used to assess mitochondrial injury across the three groups. The red fluorescence showed the normal MMP level while the green fluorescence showed a loss of MMP. As shown in Fig. 2a, there was a significantly reduced MMP level in the lung tissue of the CIH exposure group when compared to that of the NC group (p < 0.05). While the Ad supplement group (CIH + Ad) had a significantly higher MMP levels compared to the CIH group, these levels were still remarkably lower than those observed in the NC group (all p < 0.05).

MMP and ROS levels. Supplementation with Ad increased the MMP level and inhibited ROS production to counteract the effects of CIH. a The MMP level in pulmonary tissue from the three study groups. b The ROS level in pulmonary tissue from the three study groups. c, d Densitometric evaluation of MMP and ROS levels. NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars show ± SD. *p < 0.05
DHE staining was used to evaluate ROS levels in pulmonary tissue across the three groups. The red fluorescence intensity represented the ROS level. As shown in Fig. 2b, the ROS level was higher in the CIH group than in the NC group (p < 0.05). The ROS level was lower in the CIH + Ad group than in the CIH group (p < 0.05), but was higher in the CIH + Ad group than in the NC group (p < 0.05).
Mitochondria-associated apoptosis pathway
Mitochondria-associated apoptosis pathway was estimated in three groups by detecting the mitochondrial/cytoplasmic protein expression ratios of cytochrome c and bcl-2/Bax. The abnormal distribution of cytochrome C and the abnormal ratios of bcl-2/Bax proteins indicated the activation of mitochondria-associated apoptosis pathway. As shown in Fig. 3, the mitochondrial/cytoplasmic cytochrome c and bcl-2/Bax protein expression ratios were lower in the CIH group than in the NC group (all p < 0.05). However, the ratios in the CIH + Ad group were higher than those in the CIH group (all p < 0.05), but were lower than those in the NC group (all p < 0.05).

Mitochondrial injury-associated pulmonary cell apoptosis. Supplementation with Ad blocked CIH-induced mitochondrial injury-associated pulmonary cell apoptosis. a The protein levels of bcl-2 and Bax, as well as mitochondrial and cytosolic cytochrome c in pulmonary tissue in three groups. b, c Densitometric evaluation of independent Western blots. NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars denote ± SD. *p < 0.05
ER stress-associated apoptosis pathway
The activation of ER stress-associated apoptosis of pulmonary cells across the three groups was assayed by detecting the protein expressions of GRP78, CHOP, TRAF2, and the ratios of cleaved caspase-12/caspase-12, p-IRE1/IRE1, p-JNK/JNK, and p-P38/P38 MAPK. The high expressions of these proteins and protein ratios indicated activation of the ER stress-associated apoptosis pathway. As shown in Fig. 4, the protein expressions of GRP78, CHOP, TRAF2, and the protein expression ratios of cleaved caspase-12/caspase-12, p-IRE1/IRE1, p-JNK/JNK, and p-P38/P38 MAPK were all higher in the CIH group than in the NC and CIH + Ad groups (all p < 0.05). The protein expressions of CHOP, TRAF2, and the protein expression ratios of cleaved caspase-12/caspase-12, p-IRE1/IRE1, and p-JNK/JNK were also higher in the CIH + Ad group than in the NC group (all p < 0.05).

ER stress-associated pulmonary cell apoptosis. Supplementation with Ad blocked CIH-induced ER stress-associated pulmonary cell apoptosis. a The protein levels of GRP78, p-IRE1, TRAF2, p-JNK, p-P38 MAPK, CHOP, and caspase-12 in lung tissue in three groups. b–g Densitometric evaluation of independent Western blots. NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars denote ± SD. *p < 0.05
Death receptor pathway
The protein expressions of Fas-associated death domain (FADD), Fas, and FasL were assayed to evaluate activation of the death receptor pathway in pulmonary cells across the three groups. As shown in Fig. 5, the expression of these proteins was higher in the CIH group than in the NC group. The expression of these proteins in the CIH + Ad group was lower than in the CIH group, but higher than in the NC group (all p < 0.05).

Death receptor pathway-associated pulmonary cell apoptosis. Supplementation with Ad blocked CIH-induced death receptor pathway-associated pulmonary cell apoptosis. a The protein levels of Fas, FasL, and FADD in pulmonary tissue in three groups. b–d Densitometric evaluation of independent Western blots. NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars denote ± SD. *p < 0.05
SIRT3/SOD2 pathway
SOD2 is an important mitochondrial oxidative scavenger and regulator of ROS, and SIRT3 is a key regulator of SOD2 [19]. SOD2 is the deacetylated form of Ac-SOD2, which can inhibit mitochondrial oxidative stress and reduce the production of ROS [9, 19]. As shown in Fig. 6, exposure to CIH reduced the protein expression of SIRT3 and the protein expression ratio of SOD2/Ac-SOD2, when compared with the NC group (all p < 0.05). In the CIH + Ad group, Ad supplementation ameliorated the CIH-related reduction in SIRT3 expression and reduction in the SOD2/Ac-SOD2 ratio. However, SIRT3 protein expression and the protein expression rate of SOD2/Ac-SOD2 in the CIH + Ad group were still lower than in the NC group (all p < 0.05).

The SIRT3/SOD2 signaling pathway. CIH reduced the expression of SIRT3 protein and the expression ratio of SOD2/Ac-SOD2 proteins, whereas supplementation with Ad increased the expression of SIRT3 protein and the expression ratio of SOD2/Ac-SOD2 proteins. a The protein levels of Ac-SOD2, SOD2, and SIRT3 in pulmonary tissue in three groups. b, c Densitometric evaluation of independent Western blots. NC, normal control; CIH, chronic intermittent hypoxia; CIH + Ad, chronic intermittent hypoxia plus adiponectin supplement. N = 3, error bars denote ± SD. *p < 0.05
Discussion
OSAHS is a prevalent condition caused by dynamic upper airway collapse during sleep. The pathological impact and consequences are due to CIH. CIH models were widely applied to imitate the hypoxia condition in OSAHS [20]. The effects of OSAHS on pulmonary function were evaluated in a rat model of CIH. Twelve weeks of CIH exposure did not lead to decrease in pulmonary function or an increase in pulmonary arterial pressure. Fagan et al. found that CIH induced pulmonary vascular remodeling and PH in mice [6]. Our study was different from the study completed by Fagan KA with respect to the animal species used and the environment, the method of CIH-modeling. These differences might contribute to the contradictory results between the two studies. Nara reported that IH could induce PH in old rats (9 months of age), but not in young rats (2 months of age) [21], and in the present study, we used rats aged 8 weeks. However, we found that CIH induced pulmonary injuries at the molecular level, which was demonstrated by high rates of pulmonary cell apoptosis (Fig. 1), high levels of ROS (Fig. 2b), low levels of MMP (Fig. 2a), and high activation of apoptotic pathways (Figs. 3, 4, and 5). Yao found that the high pulmonary cell apoptosis and oxidative stress were participated in the lung injury induced by paraquat, and Li found that the high pulmonary cell apoptosis, high ROS level, and ER stress were upregulated in seawater inhalation-induced lung injury [22, 23]. Consistent with their study, we also observed high pulmonary cell apoptosis and ROS, which were considered as the marks of pulmonary injury induced by CIH. In our model, pulmonary cell apoptosis observed, but no changes in pulmonary function or PH were found after CIH exposure, this might indicate that early pulmonary injuries occurred at the molecular level after CIH exposure in the present study.
CIH, as a special type of hypoxia, has previously been shown to induce the production of ROS. The imbalance between oxidative and antioxidative activity is well reported to lead to serious physiological abnormalities [8, 24]. In this study, the significant elevation of ROS that occurred in pulmonary tissue after CIH exposure was consistent with previous observations that CIH increases oxidative stress in rat lung tissue [25]. Excessive ROS production is harmful and can lead to cell apoptosis [26]. In this study, increased lung cell apoptosis was found after CIH exposure. This result implied that ROS contributed to the development of pulmonary injury.
Mitochondria are the cellular centers for aerobic respiration and energy production and are also the major source of ROS production. When mitochondria are damaged, MMP levels are reduced, which could result in excessive ROS production [27]. Therefore, the decrease in MMP levels may be a potential mechanism for CIH-induced ROS. Accumulation of intracellular ROS can further damage mitochondria and activate mitochondria-associated cell apoptosis [28]. ROS induce the influx of mitochondrial cytochrome c into the cytoplasm and regulate the expression of Bcl-2 family members [29], which are involved in mitochondria-associated apoptosis [30]. In this study, excess ROS activated mitochondria-associated apoptosis. This was evident based upon the upregulation of Bax, which promoted apoptosis, the downregulation of bcl-2 and MMP levels, which inhibited apoptosis, and the abnormal distribution of cytochrome c after CIH exposure. Thus, activation of mitochondria-mediated apoptosis by ROS most likely contributed to CIH-induced lung injury.
In addition to mitochondrial apoptosis, ROS can also activate ER stress and death receptor-associated apoptosis pathways [27, 31]. Excess ROS activates apoptosis by phosphorylation of IRE1, which activates the downstream targets phospho-JNK and phospho-p38 MAPK and leads to cell apoptosis [32]. All three pathways can increase CHOP expression [33] and caspase-12 activity, which are associated with cell apoptosis [34]. In accordance with these studies, the expression levels of CHOP, GRP78, IRE1/JNK pathway proteins, and cleaved caspase-12 were increased in pulmonary tissue after CIH exposure in this study. Fas-FasL signaling was one of the death receptor pathways [35], which was activated in the CIH model rats in this study. These results suggested that mitochondria-, ER stress-, and death receptor-associated apoptosis were involved in apoptosis of lung cells induced by ROS that were generated in response to CIH.
In this study, we demonstrated that Ad partially alleviated pulmonary tissue injury, protecting lung tissue against injury induced by CIH. Ad induced the MMP level, reduced lung cell apoptosis, ROS levels, and inhibited the mitochondrial-, ER stress-, and death receptor- associated apoptosis pathways in this study. The results were consistent with previous studies by Yao et al., who found that Ad protected against lung injury induced by paraquat in mice [22] and by Sliman et al., who reported that Ad ameliorated hyperoxia-induced lung damage in mice [36]. Taken together, the available evidence indicates that Ad can protect the lung tissue from various types of injury. Previous studies have linked the mechanism of Ad protection to inhibition of ROS production in myocardial ischemic reperfusion injury, lung injury, and neutrophil injury [36,37,38,39]. Ad has also been found to inhibit ROS production in rat myocardial and kidney tissues [17, 18]. Consistent with these studies, Ad blocked CIH-induced production of ROS in lung tissue, which in turn inhibited the activation of the three apoptotic pathways and decreased pulmonary cell apoptosis. However, it remains to be determined which one of the three apoptotic pathways was primarily inhibited by Ad.
To explore how Ad reduced ROS production, we examined the protein expression involved in the SIRT3/SOD2 signaling pathway. SIRT3 is a member of the sirtuin family that controls deacetylation of downstream proteins, such as Ac-SOD2 and HIF-α. SIRT3 is located within mitochondria and regulates mitochondrial function by deacetylating Ac-SOD2. SOD2, the deacetylated form of Ac-SOD2, is an important regulator of mitochondrial ROS production and it can inhibit mitochondrial oxidative stress as well as reduce the production of ROS [9, 19]. Chen et al. found that activation of the SIRT3/SOD2 pathway reduced ROS production in response to bile acids [40]. Cheng et al. found that SIRT3/SOD2 pathway overexpression inhibited the linalool-induced increase of mitochondrial ROS production [41]. In this study, the SIRT3 protein expression and the ratio of SOD2/Ac-SOD2 were increased after Ad supplement, which indicated that Ad activates the SIRT3/SOD2 pathway and blocks CIH-induced ROS production.
Conclusions
Abnormal pulmonary function and PH were not found after 12 weeks of CIH, but CIH-associated pulmonary tissue injury was found at the molecular level. CIH exposure leads to loss of MMP, excess production of ROS, and activation of the mitochondrial-, ER stress-, and death receptor-associated apoptosis pathways. This resulted in excess pulmonary cell apoptosis, which contributed to early pulmonary tissue injury. Supplementation with Ad ameliorated this pulmonary injury by activating the SIRT3/SOD2 signaling pathway, reducing ROS production, and inhibiting ROS-associated pulmonary cell apoptosis. Further studies are needed to elucidate the detailed mechanism through which Ad exerts its protective role in vivo.
References
Feldstein CA (2016) Blood pressure effects of CPAP in nonresistant and resistant hypertension associated with OSA: a systematic review of randomized clinical trials. Clin Exp Hypertens 38(4):337–346. https://doi.org/10.3109/10641963.2016.1148156
Khan F, Walsh C, Lane SJ, Moloney E (2014) Sleep apnoea and its relationship with cardiovascular, pulmonary, metabolic and other morbidities. Ir Med J 107(1):6–8
Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiery JL (2016) Left ventricular heart failure and pulmonary hypertension. Eur Heart J 37(12):942–954. https://doi.org/10.1093/eurheartj/ehv512
Wong HS, Williams AJ, Mok Y (2017) The relationship between pulmonary hypertension and obstructive sleep apnea. Curr Opin Pulm Med 23(6):517–521. https://doi.org/10.1097/MCP.0000000000000421
Wong HT, Chee KH, Chong AW (2017) Pulmonary hypertension and echocardiogram parameters in obstructive sleep apnea. Eur Arch Otorhinolaryngol 274(6):2601–2606. https://doi.org/10.1007/s00405-017-4491-1
Fagan KA (2001) Selected contribution: pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol 90(6):2502–2507
Hamada S, Sato A, Hara-Chikuma M, Satooka H, Hasegawa K, Tanimura K, Tanizawa K, Inouchi M, Handa T, Oga T, Muro S, Mishima M, Chin K (2016) Role of mitochondrial hydrogen peroxide induced by intermittent hypoxia in airway epithelial wound repair in vitro. Exp Cell Res 344:143–151. https://doi.org/10.1016/j.yexcr.2016.04.006
Pelster B, Wood CM, Jung E, Val AL (2018) Air-breathing behavior, oxygen concentrations, and ROS defense in the swimbladders of two erythrinid fish, the facultative air-breathing jeju, and the non-air-breathing traira during normoxia, hypoxia and hyperoxia. J Comp Physiol B 188(3):437–449. https://doi.org/10.1007/s00360-017-1142-1
Katwal G, Baral D, Fan X, Weiyang H, Zhang X, Ling L, Xiong Y, Ye Q, Wang Y (2018) SIRT3 a major player in attenuation of hepatic ischemia-reperfusion injury by reducing ROS via its downstream mediators: SOD2, CYP-D, and HIF-1alpha. Oxidative Med Cell Longev 2018:2976957–2976913. https://doi.org/10.1155/2018/2976957
Ding W, Zhang Q, Dong Y, Ding N, Huang H, Zhu X, Hutchinson S, Gao X, Zhang X (2016) Adiponectin protects the rats liver against chronic intermittent hypoxia induced injury through AMP-activated protein kinase pathway. Sci Rep 6:34151. https://doi.org/10.1038/srep34151
Fisman EZ, Tenenbaum A (2014) Adiponectin: a manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc Diabetol 13:103. https://doi.org/10.1186/1475-2840-13-103
Van Berendoncks AM, Garnier A, Ventura-Clapier R, Conraads VM (2012) Adiponectin: key role and potential target to reverse energy wasting in chronic heart failure. Heart Fail Rev 18:557–566. https://doi.org/10.1007/s10741-012-9349-4
Park M, Sweeney G (2012) Direct effects of adipokines on the heart: focus on adiponectin. Heart Fail Rev 18:631–644. https://doi.org/10.1007/s10741-012-9337-8
Al Mutairi S, Mojiminiyi OA, Al Alawi A, Al Rammah T, Abdella N (2014) Study of leptin and adiponectin as disease markers in subjects with obstructive sleep apnea. Dis Markers 2014:706314–706318. https://doi.org/10.1155/2014/706314
Kim Y, Cho JY, Oh SW, Kang M, Lee SE, Jung E, Park YS, Lee J (2018) Globular adiponectin acts as a melanogenic signal in human epidermal melanocytes. Br J Dermatol 179(3):689–701. https://doi.org/10.1111/bjd.16488
Kim EH, Park PH (2018) Globular adiponectin protects rat hepatocytes against acetaminophen-induced cell death via modulation of the inflammasome activation and ER stress: critical role of autophagy induction. Biochem Pharmacol 154:278–292. https://doi.org/10.1016/j.bcp.2018.05.014
Ding W, Cai Y, Wang W, Ji L, Dong Y, Zhang X, Su M, Liu J, Lu G, Zhang X (2016) Adiponectin protects the kidney against chronic intermittent hypoxia-induced injury through inhibiting endoplasmic reticulum stress. Sleep Breath 20:1069–1074. https://doi.org/10.1007/s11325-016-1321-4
Ding W, Zhang X, Huang H, Ding N, Zhang S, Hutchinson SZ, Zhang X (2014) Adiponectin protects rat myocardium against chronic intermittent hypoxia-induced injury via inhibition of endoplasmic reticulum stress. PLoS One 9(4):e94545. https://doi.org/10.1371/journal.pone.0094545
Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D (2014) Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal 20(10):1646–1654. https://doi.org/10.1089/ars.2013.5482
Celec P, Jurkovicova I, Buchta R, Bartik I, Gardlik R, Palffy R, Mucska I, Hodosy J (2013) Antioxidant vitamins prevent oxidative and carbonyl stress in an animal model of obstructive sleep apnea. Sleep Breath 17(2):867–871. https://doi.org/10.1007/s11325-012-0728-9
Nara A, Nagai H, Shintani-Ishida K, Ogura S, Shimosawa T, Kuwahira I, Shirai M, Yoshida K (2015) Pulmonary arterial hypertension in rats due to age-related arginase activation in intermittent hypoxia. Am J Respir Cell Mol Biol 53(2):184–192. https://doi.org/10.1165/rcmb.2014-0163OC
Yao R, Zhou Y, He Y, Jiang Y, Liu P, Ye L, Zheng Z, Lau WB, Cao Y, Zeng Z (2015) Adiponectin protects against paraquat-induced lung injury by attenuating oxidative/nitrative stress. Exp Ther Med 9(1):131–136. https://doi.org/10.3892/etm.2014.2073
Li PC, Wang BR, Li CC, Lu X, Qian WS, Li YJ, Jin FG, Mu DG (2018) Seawater inhalation induces acute lung injury via ROS generation and the endoplasmic reticulum stress pathway. Int J Mol Med 41(5):2505–2516. https://doi.org/10.3892/ijmm.2018.3486
Xu Y, Wang W, Jin K, Zhu Q, Lin H, Xie M, Wang D (2017) Perillyl alcohol protects human renal tubular epithelial cells from hypoxia/reoxygenation injury via inhibition of ROS, endoplasmic reticulum stress and activation of PI3K/Akt/eNOS pathway. Biomed Pharmacother 95:662–669. https://doi.org/10.1016/j.biopha.2017.08.129
Kou Y, Zhang P, Wang H, Zhang J, Han X, Yu J, Wang L, Zhang M (2015) Changes of angiotensin II and oxidation stress during the development of chronic intermittent-induced pulmonary injury in rats. Zhonghua Jie He He Hu Xi Za Zhi 38(8):612–616
Liu W, Yang T, Xu Z, Xu B, Deng Y (2019) Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free Radic Res 53(1):26–44. https://doi.org/10.1080/10715762.2018.1546852
Liu XR, Li T, Cao L, Yu YY, Chen LL, Fan XH, Yang BB, Tan XQ (2018) Dexmedetomidine attenuates H2O2-induced neonatal rat cardiomyocytes apoptosis through mitochondria- and ER-medicated oxidative stress pathways. Mol Med Rep 17(5):7258–7264. https://doi.org/10.3892/mmr.2018.8751
Ma Y, Zhang J, Zhang Q, Chen P, Song J, Yu S, Liu H, Liu F, Song C, Yang D, Liu J (2014) Adenosine induces apoptosis in human liver cancer cells through ROS production and mitochondrial dysfunction. Biochem Biophys Res Commun 448(1):8–14. https://doi.org/10.1016/j.bbrc.2014.04.007
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101. https://doi.org/10.1016/j.devcel.2011.06.017
Kalashnikova I, Mazar J, Neal CJ, Rosado AL, Das S, Westmoreland TJ, Seal S (2017) Nanoparticle delivery of curcumin induces cellular hypoxia and ROS-mediated apoptosis via modulation of Bcl-2/Bax in human neuroblastoma. Nanoscale 9(29):10375–10387. https://doi.org/10.1039/c7nr02770b
Nebbioso A, Pereira R, Khanwalkar H, Matarese F, Garcia-Rodriguez J, Miceli M, Logie C, Kedinger V, Ferrara F, Stunnenberg HG, de Lera AR, Gronemeyer H, Altucci L (2011) Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Mol Cancer Ther 10(12):2394–2404. https://doi.org/10.1158/1535-7163.MCT-11-0525
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115(10):2656–2664. https://doi.org/10.1172/JCI26373
Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell 24:2477–2490. https://doi.org/10.1091/mbc.E13-01-0067
Li XF, Zhang Z, Chen ZK, Cui ZW, Zhang HN (2018) [Retracted] Piezo1 protein induces the apoptosis of human osteoarthritisderived chondrocytes by activating caspase12, the signaling marker of ER stress. Int J Mol Med 42(6):3640. https://doi.org/10.3892/ijmm.2018.3918
Luo Q, Yang D, Qi Q, Huang C, Chen B, Liu W, Shi L, Xia Y, Tang L, Fang J, Ou Y, Geng Y, Chen Z (2018) Role of the death receptor and endoplasmic reticulum stress signaling pathways in polyphyllin I-regulated apoptosis of human hepatocellular carcinoma HepG2 cells. Biomed Res Int 2018:5241941–5241911. https://doi.org/10.1155/2018/5241941
Sliman SM, Patel RB, Cruff JP, Kotha SR, Newland CA, Schrader CA, Sherwani SI, Gurney TO, Magalang UJ, Parinandi NL (2013) Adiponectin protects against hyperoxic lung injury and vascular leak. Cell Biochem Biophys 67(2):399–414. https://doi.org/10.1007/s12013-011-9330-1
Goldstein BJ, Scalia RG, Ma XL (2009) Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 6(1):27–35. https://doi.org/10.1038/ncpcardio1398
Magalang UJ, Rajappan R, Hunter MG, Kutala VK, Kuppusamy P, Wewers MD, Marsh CB, Parinandi NL (2006) Adiponectin inhibits superoxide generation by human neutrophils. Antioxid Redox Signal 8(11–12):2179–2186. https://doi.org/10.1089/ars.2006.8.2179
Potenza MA, Sgarra L, Nacci C, Leo V, De Salvia MA, Montagnani M (2019) Activation of AMPK/SIRT1 axis is required for adiponectin-mediated preconditioning on myocardial ischemia-reperfusion (I/R) injury in rats. PLoS One 14(1):e0210654. https://doi.org/10.1371/journal.pone.0210654
Chen Y, Qing W, Sun M, Lv L, Guo D, Jiang Y (2015) Melatonin protects hepatocytes against bile acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radic Res 49(10):1275–1284. https://doi.org/10.3109/10715762.2015.1067806
Cheng Y, Dai C, Zhang J (2017) SIRT3-SOD2-ROS pathway is involved in linalool-induced glioma cell apoptotic death. Acta Biochim Pol 64(2):343–350. https://doi.org/10.18388/abp.2016_1438
Acknowledgments
We are grateful to the principal investigators of the Department of Cardiology lab.
Funding
This study was funded by the National Natural Science Foundation of China (81500069, 81700090, and 81600066) and Cooperative Research Fund of Southeast University and Nanjing Medical University (2242017K3DN44). The sponsor had no role in the design or conduct of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study was approved by the Animal Ethics Committee of Southeast University. All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ding, W., Zhang, X., Zhang, Q. et al. Adiponectin ameliorates lung injury induced by intermittent hypoxia through inhibition of ROS-associated pulmonary cell apoptosis. Sleep Breath 25, 459–470 (2021). https://doi.org/10.1007/s11325-020-02103-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11325-020-02103-3