
Abstract
Streptococcus intermedius, S. constellatus, and S. anginosus comprise the Streptococcus Milleri/Anginosus group (SMG). They are facultative anaerobic bacteria that asymptomatically colonize the upper respiratory, gastrointestinal and urogenital tracts. They are also common pathogens in pyogenic invasive infections, as well as pulmonary and urinary tract infections. Most SMG infections are polymicrobial and associated with co-infecting obligate anaerobic bacteria. To better understand the effect of oxygen on the growth and physiology of these organisms, we compared the global metabolomic and transcriptomic profiles of S. intermedius strain B196 under aerobic and anaerobic conditions. The largest transcriptional changes were associated with induction of oxidative stress response genes under aerobic conditions. Modest changes in expression of genes associated with primary metabolism were observed under the two conditions. Intracellular and extracellular metabolites were measured using HILIC–LCMS. Differences in the abundance of specific metabolites were correlated with observed transcription changes in genes associated with their metabolism, implying that metabolism is primarily regulated at the transcriptional level. Rather than a large shift in primary metabolism under anaerobic conditions our results suggest a modest tuning of metabolism to support the accelerated growth rate of S. intermedius strain B196 in the absence of oxygen. For example, under anaerobic conditions, purine metabolism, pyrimidine de novo synthesis and pyrimidine salvage pathways were up-regulated at metabolic and transcriptional levels. This study provides a better understanding of differences between S. intermedius anaerobic and aerobic metabolism. The results reflect the organism’s predilection for anaerobic growth consistent with its pathogenic association with anaerobes in polymicrobial infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Streptococcus Milleri/Anginosus group (SMG) is comprised of three distinct but closely related species of facultative anaerobic Gram-positive bacteria (S. anginosus, S. constellatus, and S. intermedius) (Gossling 1988). The SMG are often considered as commensal human microbiota and can be found asymptomatically colonizing the oral cavity, upper respiratory tract, urogenital tract and gastrointestinal tract in healthy individuals (Gossling 1988; Whiley et al. 1992). However, the SMG are also recognized pathogens in pyogenic infections including soft tissue abscesses, pleural empyema, brain and liver abscesses, and respiratory infections (Ruoff 1988; Whiley et al. 1992; Coman et al. 1995; Shinzato and Saito 1995; Laupland et al. 2006; Ripley et al. 2006; Parkins et al. 2008; Sibley et al. 2008; Siegman-Igra et al. 2012; Asam and Spellerberg 2014). Phenotypic heterogeneity in this group can make their identification challenging and recent studies suggest that the SMG are under appreciated pathogens with incidence rates for pyogenic infections comparable to Group A and Group B Streptococcus combined (Laupland et al. 2006; Siegman-Igra et al. 2012). The SMG have been primarily associated with adults with respect to both carriage and infection, however they may be underestimated in pediatric disease (Lee et al. 2010).
Most infections associated with the SMG are polymicrobial with a significant burden of obligate anaerobic bacteria present in the infection site. This has been observed in lower airway infections (Shinzato and Saito 1995; Parkins et al. 2008; Sibley et al. 2008; Filkins et al. 2012), pleural empyema (Hocken and Dussek 1985; Van der Auwera 1985; Wong et al. 1995; Sibley et al. 2012) and abscesses (Gossling 1988; Shinzato and Saito 1994; Hirai et al. 2005; Sibley et al. 2012). Understanding how SMG adapts to aerobic and anaerobic environments may provide insight into the mechanisms used by S. intermedius for survival and persistence in the host during colonization and disease progression.
In this study we examined the in vitro growth of S. intermedius strain B196 in aerobic (5 % CO2) and anaerobic (90 % N2, 5 % CO2, 5 % H2) conditions using growth kinetics, transcriptomics (RNA-seq), and both intracellular and extracellular metabolomics. The effect of oxygen on the growth, physiology and metabolism of S. intermedius provides insight in understanding its pathogenic association with anaerobes in polymicrobial infections. Moreover, understanding the metabolic regulation of S. intermedius under various oxygenated environment through comprehensive metabolomic studies can provide insights modulating its commensal or pathogenic activities in human host.
2 Materials and methods
2.1 Chemicals
HPLC grade methanol (MeOH), ethanol (EtOH), acetonitrile (ACN), and water (H2O) were purchased from Caledon laboratories (Georgetown, ON, Canada). Ammonium acetate and formic acid were purchased from Fisher Scientific Company (Ottawa, ON, Canada). 2.0 mm steel chrome ball bearings were purchased from Bearing & Oil Seals Specialists Inc. (Hamilton, ON, Canada). The isotopically labelled standards for recovery determination (RS) and for peak intensity normalization (IS) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA). Lipid standards were purchased from Avanti® Polar Lipids, Inc. (Alabaster, AL, USA), and other chemical standards for LC–MS were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Biolog Inc. (Hayward, CA, USA).
2.2 Bacterial strain, media and growth conditions
Streptococcus intermedius B196 is an invasive isolate from the hip abscess of a cystic fibrosis patient. A complete genome sequence is available for this strain (Olson et al. 2013). S. intermedius was grown on Todd Hewitt agar supplemented with 0.5 % yeast extract (THY) at 37 °C in a 5 % CO2 incubator for 3 days. A single colony was inoculated into 5 mL THY broth for overnight static growth under the above conditions. For growth kinetics, overnight cultures were inoculated into 5 mL THY broth at an initial OD600nm of 0.05 and cultured aerobically (5 % CO2) and anaerobically (90 % N2, 5 % CO2, 5 % H2) with optical density as well as colony forming units (CFU) recorded every hour (detailed procedures included in Supplementary Material ESM 1 1.1). For RNA-seq and metabolomics, the same overnight cultures were inoculated into THY broth at an initial OD600nm of 0.1 and grown under either aerobic and anaerobic conditions with samples collected at mid-exponential phase (OD600nm = 0.7).
2.3 Strand-specific RNA-seq
Three biological replicates were prepared for S. intermedius under aerobic and anaerobic conditions. A 2 mL culture from each replicate at OD600nm = 0.7 was centrifuged. The cell pellets were collected and stored in RNAprotect bacteria reagent (Qiagen, Venlo, Netherlands) for later use at −80 °C. Total cellular RNA was isolated and purified using TRIzol (Invitrogen, Carlsbad, CA, USA) and RNeasy Mini Kit (Qiagen, Venlo, Netherlands). Ribosomal RNA (rRNA) was depleted using Ribo-Zero rRNA removal Kit for bacteria (Epicentre, Madison, WI, USA). cDNA was prepared using the Superscript III first strand cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA). Strand specific RNA sequencing libraries were prepared using the dUTP approach (Parkhomchuk et al. 2009). The NEB-Next library preparation modules for Illumina were used for library preparation with a separate index used per biological replicate. The libraries were submitted to the McMaster Genomics Facility (McMaster University, Hamilton, Canada) for quality control (QC) and sequencing using standard Illumina protocol (Illumina HiSeq 1000, San Diego). QC included assessment of fragment size using BioAnalyzer and routine qPCR quantification to quantify the amount of cDNA. The libraries were converted to FastQ format using Illumina’s Casava software (version 1.8.2) with no index mismatches during demultiplexing. Approximately 20 million reads were obtained per sample with 16–17 million reads per biological replicate mapping back to the genome of S. intermedius B196 with high stringency. A detailed procedure can be found in Supplementary Material ESM 1 1.2.
2.4 RNA-seq data analysis
The transcriptomic data were aligned, assembled, analyzed and graphed using the Bowtie2: Tophat2: Cufflinks: Cuffdiff: CummeRbund pipeline (Trapnell et al. 2012). A total of 1815 genes were analyzed. Differential gene expression analysis was done using Fragments per kb per million (FPKM) values generated using the pipeline. This value takes into consideration the number of reads mapping back to each gene and normalizes that to the total number of reads generated from the sequencing run. Statistical analysis was included in the Cuffdiff analysis. Genes were classified into pathways based on BioCyc database (Caspi et al. 2014). The transcriptomic (RNA-seq) data is summarized in Table S1 and the dataset is included in Supplementary Material ESM 2.
2.5 Extraction protocol for intracellular and extracellular metabolites
The extraction procedures for intracellular and extracellular metabolomics were based upon previously published work (Fei et al. 2014). Cells from a 2 mL cell culture at 0.7 OD600nm were pelleted by centrifugation at 4 °C, washed once with 1 mL PBS for intracellular metabolomic analysis; from the same culture, 20 μL of the culture supernatant after centrifugation was collected for extracellular metabolomic analysis. Prior to extraction, 10 μL RS consisting of 770 µM l-methionine-d3 and 378 µM l-tryptophan-d5 were added to the cell pellet and THY supernatant. For intracellular metabolomics, the cell pellet was extracted with 100 μL cold methanol/ethanol/water (MeOH/EtOH/H2O, 2:2:1) and two 2.0 mm chrome steel beads using the Powerlyzer 24 (MO BIO Laboratories Inc., Carlsbad) for 2 min. The cell extract supernatant was collected after centrifugation at 9500×g for 3 min. The cell debris (consisting of precipitated protein and particulates) was extracted with 50 μL MeOH/EtOH/H2O two more times, under the same condition. For extracellular metabolomics, 20 μL THY supernatant was extracted with 80 μL MeOH/EtOH (1:1). The solution mixtures were vortex mixed for 2 min and centrifuged at 9500×g for 3 min. The clear supernatants were collected and diluted with 100 μL 60 % v/v ACN/H2O. IS with 252 μM l-phenylalanine-d8, 151 μM glycine-phenylalanine, and 88 μM diphenylalanine were added to the total 150 μL cell extracts and 200 μL supernatant extracts. Five separate controls for THY medium were also extracted as above. Both intra- and extracellular extracts were stored in −80 °C during extraction processes and before LC–MS analyses. Five biological replicates were collected for aerobic and anaerobic growth conditions and LC–MS was performed in sextuplicate for each sample.
2.6 HILIC–TOF–MS analysis for intracellular and extracellular metabolites
The HILIC–TOF–MS method and parameters were based upon previously published work (Fei et al. 2014). The intracellular and extracellular extracts were analyzed in two separate batches using an Agilent Technologies 1200 RR Series II liquid chromatograph (LC) coupled to a Bruker MicrOTOF II Mass Spectrometer. A 2 μL injection was separated on a 50 mm × 2.1 mm Kinetex 2.6 μm HILIC column of pore size of 100 Å (Phenomenex, CA, USA). The column temperature was maintained at 40 °C, and the auto sampler storage tray was set at 4 °C. The mobile phases were acetonitrile (A) and 10 mM ammonium acetate in HPLC grade water adjusted to pH 3 with formic acid (B). The flow rate was kept at 0.2 mL/min during a 24-min run with the following gradient: 95 % A for 0.5 min to 35 % A at 12.5 min with an extra 0.5 min hold, then to 95 % A at 14 min. The column was equilibrated at 95 % A for 10 min before the next injection.
The extracts were analyzed in both ESI+ and ESI− modes. The samples were acquired in random order. A quality control pooled sample was prepared by combining 5 μL extracts from all samples in either the intracellular or extracellular extract batches. The pooled samples were injected seven times at the beginning of each analysis and also after every 5 samples. MeOH/EtOH/H2O blank and a standard mixture containing IS and RS were also run after every 10 samples.
2.7 LCMS data analysis and metabolite identification
The data processing and analysis were modified from a previously published protocol (Fei et al. 2014). Post-acquisition internal calibration using intracellular sodium formate clusters in both ESI+ and ESI− were performed with Bruker’s DataAnalysis 4.0 SP4. The LC–MS data files were converted to.mzXML format using Bruker CompassXport. The metabolic features were extracted and aligned using open source XCMS with centWave algorithm (Smith et al. 2006); adducts, isotopic ions, and in-source fragments were identified using CAMERA (Kuhl et al. 2015).
To get the final metabolite feature list, metabolite features with apparent retention factor (kapp′) lower than 0.7 were removed. Isotopic ions, features resulting from IS, RS, and sodium formate clusters were also removed. The metabolite features were normalized with IS eluted closest to their retention time (i.e. features eluted before 7.40 min were normalized by phe-phe; features eluted between 7.40 and 8.30 min were normalized by l-phenylalanine-d8; features eluted after 8.30 min were normalized by gly-phe). Features with greater than 20 % variance in the pooled sample were removed to get the final metabolite feature list.
Metabolite features were identified based on accurate mass and retention time of authentic standards or compound analogs (for lipid identification only) with two identification points (Creek et al. 2014). There were 105 metabolites identified from 1885 intracellular metabolic features, 66 metabolites were identified with level 1 metabolomics standard initiative (MSI) (Sumner et al. 2007), 10 metabolites were putatively annotated with level 2 MSI, and 29 metabolites were assigned to compound classes with level 3 MSI. There were 26 phospholipids (PLs) identified to two phosphatidylcholines (PCs), one phosphatidylethanolamine (PEs), 20 phosphatidylglycerols (PGs), and three lyso-PGs in the intracellular metabolome based on the accurate mass and retention time of lipid analogs (Zheng et al. 2010; Fei et al. 2014). Similarly, there were 116 metabolites identified from 3382 extracellular metabolite features, with 82, 8 and 2 metabolites identified with level 1, 2, and 3 MSI, respectively. Both intra- and extracellular metabolomic data were summarized in Table S1 and the datasets are included in Supplementary Material ESM 3 and 4, respectively.
2.8 Multivariate statistical analyses
Both transcriptomic and metabolomic data were subjected to principal component analysis (PCA) and orthogonal partial least-squares discriminative analysis (OPLS-DA) after pareto scaling using SIMCA-P + 12.0.1 (Umetrics, Kinnelon, NJ, USA). The metabolomic data, 79 and 92 identified metabolites from S. intermedius intracellular and extracellular metabolome respectively, were also analyzed using MetaboAnalyst 3.0 for pathway analysis (default setting) based on the Staphylococcus aureus metabolic pathway (Xia et al. 2015) (Supplementary Material ESM 5). Intracellular and extracellular metabolite features and genes were assessed by univariate analyses such as Student’s t test (two-tailed, unpaired heteroscedastic) and one-way ANOVA using Microsoft Excel 2010 and MetaboAnalyst 2.0, respectively. Metabolic features and genes with p value less than 0.05 (from Student’s t test or one-way ANOVA) and fold change greater than 1.5 between conditions were considered significantly differentiated. The pathway maps were constructed based on BioCyc data for Streptococcus intermedius strains B196 and JTH08.
3 Results and discussion
3.1 Streptococcus intermedius metabolism is affected by aerobic or anaerobic growth conditions
The intracellular and extracellular metabolomes and the transcriptome were used to characterize and differentiate the responses of S. intermedius to anaerobic and aerobic growth conditions. Comprehensive multivariate OPLS-DA analyses were conducted on the 1885 intracellular and 3382 extracellular metabolic features of S. intermedius, which included polar metabolites and phospholipids. There were robust metabolomic differences between aerobic and anaerobic growth environments (Fig. 1b, c) with prediction statistic (Q2) above 0.85 (Broadhurst and Kell 2006). The extracellular metabolomic profiles of S. intermedius grown in aerobic or anaerobic conditions were distinctly different from each other, and the THY medium control, implying distinct nutrient consumption and metabolite release under these growth conditions. Twenty-eight major metabolic pathways were found using MetaboAnalyst based on identified metabolites (Fig. 2).

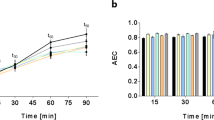
(a) The growth curves of S. intermedius cultured in either aerobic (in orange) or anaerobic (in blue) environment. The doubling time in aerobic condition was 54 min and the doubling time in anaerobic condition was 41 min. The optical density of the cells was measured every hour in triplicate. OPLS-DA score plots summarizing (b) 1885 metabolite features found in the intracellular extracts between aerobically and anaerobically cultured S. intermedius, (c) 3382 metabolite features found in extracellular medium of aerobically and anaerobically cultured S. intermedius and Todd Hewitt growth media, and (d) gene expression differences including 1815 transcripts obtained from RNA-seq of S. intermedius grown in either aerobic or anaerobic conditions. The samples belonging to the same treatment were highlighted with circles (Color figure online)

A heatmap of 28 pathways and how they were affected by aerobic or anaerobic growth conditions, based on identified metabolites in the intracellular and extracellular metabolomes of S. intermedius. The p value of each pathway was computed by pathway analysis using MetaboAnalyst 2.0 using Gram-positive Staphylococcus aureus as the model organism. Rows were metabolite pathways; columns were comparisons between treatments including aerobic and anaerobic growth conditions and the Todd Hewitt media blank. Comparisons were based on either intracellular S. intermedius cell extracts of 79 identified metabolites (exclude phospholipids) or extracellular supernatant of 92 identified metabolites. The color key indicates the −log10 of p values for pathway significance (refer to the color scale). The energy production pathways were calculated mostly based on C5, C6 monosaccharide and disaccharide abundances. A list of identified metabolites used for the analyses was included in the supplementary material.*S. intermedius does not have the tricarboxylic acid cycle (TCA); the TCA pathway shown here was based on Staphylococcus aureus metabolism (Color figure online)
The presence of oxygen affected both the intracellular and extracellular metabolome of S. intermedius. Over 37.7 % of the intracellular metabolite features (710/1885 metabolite features) were differentially produced when comparing aerobic and anaerobic growth environments. Among these, 327 features were more abundant under anaerobic condition, and 383 features were less abundant (Fig. S1a). Similarly, 38.6 % (1307/3382 features) of the extracellular metabolite features were significantly different among aerobic, anaerobic supernatants and THY medium controls. Although the majority of changes occurred between THY and growth conditions, 3.4 % (114/3382) of the features were differentially expressed between aerobic and anaerobic supernatants (28 increased and 86 decreased under anaerobic growth conditions (Fig. S1b-d).
The RNA-seq dataset of S. intermedius B196 captured the transcription of 1815 genes based on the current annotation (Olson et al. 2013). These genes were classified to 58 gene pathways. The transcription profiles under aerobic and anaerobic growth were distinct as shown in OPLS-DA score plot (Fig. 1d). There were 625 genes that significantly affected with p values ≤10−4.5 (the Cuffdiff threshold cut-off) (Trapnell et al. 2012). Of these, 297 genes had greater than twofold changes in gene expression (Fig. S2). Figure 3 illustrates the percentage of genes in each pathway that were up-regulated or down-regulated with a change greater than twofold. The pathways can be divided into four groups based on the responses seen: (1) Aerobic Response only, where genes in pathway were up-regulated only in the presence of oxygen (2) Anaerobic Response only, where genes were up-regulated in the absence of oxygen, (3) Mixed Response, which includes a subset of genes up-regulated and a subset down-regulated under each condition, and (4) An unaffected group, where there was no oxygen dependent response. The data confirms that aerobic and anaerobic growth conditions can lead to global metabolic and transcriptional changes in S. intermedius.

The effect of the presence of oxygen on gene expression of S. intermedius. Anaerobic gene expression was compared to aerobic and visualized with down-regulated, upregulated and unaffected genes coloured in orange, yellow and green respectively. Genes were classified into pathways and assembled into 4 groups based on response, namely, anaerobic response, mixed response, aerobic response and unaffected pathways. CRISPR clustered regularly interspaced short palindromic repeats, SAM S-adenosyl methionine, PPP pentose phosphate pathway, CHP conserved hypothetical protein, GlcNAc N-acetylglucosamine, FMN flavin mononucleotide, UDP uridine diphosphate, NAD nicotinamide adenine dinucleotide (Color figure online)
3.2 Adaptations affecting growth
Streptococcus intermedius exhibited differential growth kinetics in the presence and absence of oxygen. Under aerobic conditions, S. intermedius exhibited an extended lag phase and slower overall growth rate compared to anaerobic growth conditions (Fig. 1a, S3). The doubling time during the logarithmic phase was 41 min under anaerobic conditions and 54 min under aerobic conditions based on CFUs (Fig. S3). Consistent with the increased growth rate under anaerobic conditions, pathways associated with central carbon metabolism, the arginine deaminase pathway, pyrimidine and purine metabolism were found to be increased under anaerobic conditions using both transcriptomics and metabolomics.
3.2.1 Central carbon metabolism of S. intermedius was up-regulated during anaerobic growth
Many facultative anaerobes such as E. coli, can adapt to different oxygen environments by switching from aerobic to anaerobic respiration or fermentative metabolism under oxygen-deficient conditions (Trotter et al. 2011). In aerobic respiration, oxygen is used as the terminal electron acceptor in the electron transport chain, which generates the proton gradient across the cell membrane and allows ATP to be generated by the cell. Lactic acid bacteria, including SMG, lack heme, the main component of the cytochromes in the electron transport chain. To compensate, lactic acid bacteria generate NAD+ and acidic byproducts (lactate, acetate and formate) via mixed acid fermentation (Crow and Pritchard 1977). This occurs under both aerobic and anaerobic conditions. The acidic byproducts are exported and generate a proton gradient across the cell membrane, allowing ATP synthesis. Here, the effect of oxygen on glycolysis and mixed acid fermentation in S. intermedius were analyzed by transcriptomics and metabolomics (Fig. 4).

Overview of a glycolysis and mixed acid fermentation pathways and b NAD+/NADH cycling pathway of S. intermedius, which were affected by aerobic and anaerobic growth conditions. Genes that were up-regulated in anaerobic conditions were indicated in blue; genes that were down-regulated in anaerobic condition were indicated in red; genes which expression was not statistically significant were indicated in black (Student’s t test, p > 0.05). PTS phosphotransferase system, manL putative phosphotransferase system, mannose-specific EIIAB; manM, PTS system, mannose-specific IIC component; manN, PTS system, mannose-specific IID component; glcK, glucokinase putative; fba, fructose biphosphatealdolase; pyK, pyruvate kinase; pgm, putative phosphoglucomutase/phosphomannomutase; glgA, glycogen synthase, ADP-glucose type; glgB, glycogen branching enzyme; glgC, glucose-1-phosphate adenylyltransferase; glgD, glucose-1-phosphate adenylyltransferase, GlgD subunit; ldh, l-lactate dehydrogenase; pfl, formate C-acetyltransferase; pta, phosphate acetyl/butaryltransferase; ackA, acetate kinase; adhE, bifunctional acetaldehyde-CoA/alcohol dehydrogenase; adhP, alcohol dehydrogenase; aldB, alpha-acetolactate decarboxylase; ppc, phosphoenolpyruvate carboxylase; mleS, malate dehydrogenase; nox, NADH oxidase; ahpC, alkyl hydroperoxide reductase subunit C; ahpF, alkyl hydroperoxide reductase subunit F (Color figure online)
Genes associated with glycolysis including glucokinase (glcK, 2.83-fold) and fructose 1,6-bisphosphate aldolase (fba, 2.69-fold), were up-regulated under anaerobic conditions (Fig. 4a). Additionally, expression of the glycogen biosynthesis operon (glgABCD) was also increased by more than twofold under anaerobic growth, implying that glucose utilization exceeds energy requirements and is therefore stored in the form of glycogen. There was also a decreased expression of genes involved in the synthesis of acetoin, namely acetolactate synthase (SIR_RS12085, 5.32-fold) and aldehyde dehydrogenase (aldB, 6.13-fold) as has been found in other lactic acid bacteria such as Lactococcus lactis (Bassit et al. 1993). Regulation of enzymatic activity by glycolytic intermediates and other metabolites could alter affect carbohydrate metabolism and contribute to the increased growth kinetics observed under anaerobic conditions. This will not be reflected in the transcriptional profile.
The presence of oxygen in the growth environment is known to affect genes associated with carbohydrate uptake in non-SMG streptococci (Ahn et al. 2007, 2009). The utilization of C5 and C6 sugars (e.g. ribose, glucose, trehalose) from the extracellular medium was equivalent between growth conditions. However, eight genes involved in fructose, ascorbate, glucose, mannose and N-acetylgalactosamine transport were up-regulated under anaerobic conditions (Fig. S4). Conversely, the expression of seven genes involved in the uptake of trehalose, lactose, starch and glycerol as well as six putative carbohydrate uptake genes were down-regulated during anaerobic growth. These data suggest that regulation of carbohydrate uptake is also a feature of S. intermedius metabolism when grown under different oxygen levels.
The NAD+/NADH cycling pathway and carbohydrate metabolism are inextricably linked in S. intermedius (Fig. 4b). Under anaerobic conditions, lactate dehydrogenase (ldh, 3.30-fold) and malate dehydrogenase (mleS, 2.31-fold) were up-regulated with glycolysis genes to allow regeneration of NAD+ (Fig. 4b, S4). However, under aerobic conditions, nox and ahpCF are up-regulated while ldh and mleS are down-regulated, implying a change in the mechanism of NAD+/NADH cycling. Along with a lower expression of glycolysis genes under aerobic conditions, these two features may contribute to slower growth.
3.2.2 Up-regulation of arginine deiminase pathway in an anaerobic growth environment could lead to increase in de novo synthesis of pyrimidine
In host-pathogen interactions, the ability of bacteria to compete for nutrients with host cells is essential for bacterial colonization and pathogenesis. Amino acids have been used as the primary carbon source by bacteria in rich media (Prüb et al. 1994; Sezonov et al. 2007). However, we observed minimal net change in amino acid concentrations while comparing the growth conditions to the original THY medium aside from arginine (Fig. S5). Thus oxygen had little impact on the metabolism of other amino acids.
Arginine is known to be required for optimal SMG growth (Rogers et al. 1987) and was consumed during S. intermedius growth in our experiments. The arginine deiminase (ADI) pathway is used for energy production and also feeds into de novo synthesis of pyrimidine via carbamoyl phosphate (Zúñiga et al. 2002; Gruening et al. 2006; Cusumano and Caparon 2015). There are four enzymes involved in the arginine deiminase (ADI) pathway: arginine deiminase (arcA), ornithine carbamoyltransferase (arcB), carbamate kinase (arcC) and arginine/ornithine antiporter (arcD) (Gupta et al. 2013) (Fig. 5). The production of ammonia and ATP from carbamoyl phosphate via ArcC provides energy and protection against acid stress (Marquis et al. 1987; Cotter and Hill 2003).The gene expression of arcC was similar under both growth conditions Therefore, the energy generation or acid stress resistance provided by carbamoyl phosphate and ADI is comparable under both conditions. On the other hand, the expression of arcA, arcB and arcD, which lead to the synthesis of carbamoyl phosphate, were all increased significantly under anaerobic growth conditions. Additionally, the intracellular concentrations of arginine and ornithine (the carbamoyl phosphate by-product) were also elevated in the anaerobic conditions. Overall, this implies that under anaerobic condition, there is a greater conversion of arginine to carbamoyl phosphate for de novo synthesis of pyrimidine via the ADI pathway. Though the overall consumption of arginine was slightly greater in aerobic conditions, the arginine was preferentially metabolized to citrulline and exported, which resulted a three-fold greater extracellular citrulline concentration. Thus, up-regulation of the ADI pathway under anaerobic conditions contributed to the increased production of carbamoyl phosphate and consequently, the up-regulation of pyrimidine de novo synthesis. Many pathogens use the consumption of arginine as a means to impair the host’s ability to produce nitric oxide (Stadelmann et al. 2013; Cusumano et al. 2014) and this may be a strategy used by S. intermedius in infections.

The arginine deiminase and pyrimidine de novo synthesis pathways. Pathway was constructed based on the BioCyc database for S. intermedius B196 and JTH08. The metabolite names were written in black and the gene names were written in green. The fold changes of metabolite expression were indicated in color scaled boxes for S. intermedius grown in aerobic (A) and anaerobic (AN) conditions and the Todd Hewitt media blank (TH). The endo-metabolome was colored in grey and the exo-metabolome was light green, fold changes in gene expressions were indicated by numerical values. The undetected metabolites were indicated with a black filled box. The fold changes in metabolite or gene levels were calculated respective to aerobic growth conditions for either endo- or exo-extracts, where an increase was shown in blue and a decrease was shown in red. n = 7 except for intracellular cell extract in aerobic conditions and Todd Hewitt media. *p < 0.05, **p < 0.005, ***p < 0.0001 (Color figure online)
3.2.3 Anaerobic growth conditions enhance pyrimidine and purine metabolism in S. intermedius
The intracellular concentrations of cytosine, cytidine, adenosine, adenine, guanosine, and uridine were all higher during anaerobic growth, while their extracellular abundances were much lower implying greater cellular uptake in comparison to the aerobic culture (Fig. S6, S7). In agreement with the metabolic data, the salvage and de novo nucleotide synthesis pathways were also both up-regulated anaerobically at the transcription level
The pyrimidine de novo synthesis pathway was elevated during anaerobic growth. This was indicated by the increase of intracellular levels of orotate, a pathway intermediate, and the increased expression of pyrimidine synthesis genes (pyrBCDEF) (Fig. 5, S8). Pathways for production of carbamoyl phosphate (ADI pathway), aspartate (aspartate aminotransferase), and bicarbonate (carAB), precursors for pyrimidine de novo synthesis, were all up-regulated anaerobically. Moreover, the purine metabolism pathways were also elevated anaerobically. Intracellular adenine level had shown great disparity according to the aerobic and anaerobic growth condition. Compared to aerobic condition, more than threefold increase had been observed for intracellular adenine when S. intermedius was anaerobically cultured. Though the adenine level in the spent media was reduced in both growth conditions, a greater reduction was noted in the anaerobic growth. It may suggest greater influx of adenine under anaerobic growth. The genes involved in the inter-conversion between nucleosides and nucleotides were also up-regulated anaerobically to adjust to the high demand of intracellular metabolites (Fig. S6, S7). The enhanced expression of nucleoside and nucleotide metabolism genes may contribute to the increased growth rate of S. intermedius under anaerobic conditions.
3.3 Adaptation to oxidative stress
Protection against oxidative stress from both internally produced and exogenous reactive oxygen species is important for the Streptococci (Higuchi et al. 2000; Jakubovics et al. 2002). Oxidative stress can cause damage to iron-sulfur cluster containing proteins as well as DNA (Imlay 2013). Genes involved in oxidative stress response were the most differentially expressed genes in our study. Under aerobic conditions, NADH oxidase (nox, 7.38-fold) was up-regulated in comparison to anaerobic conditions, as was the the alkyl-hydroperoxidase system (ahpCF, 25.52- and 28.35-fold), the peroxide resistance protein (dps; 5.74-fold) and superoxide dismutase (sodA; 4.96-fold). Dps removes free iron from the cell, preventing the generation of peroxides and SodA degrades superoxides while generating hydrogen peroxide which can then be reduced to water by ahpCF via NAD+/NADH cycling pathway. These adaptations to oxidative stress have been observed in S. mutans (Higuchi 1984; Higuchi et al. 2000; Ahn et al. 2007).
Redox balance is important for cell homeostasis, and it is in part maintained through NAD+/NADH cycling pathways, where NADH is oxidized to NAD+ for glycolysis (Fig. 4b). We also observed that under aerobic conditions, genes involved in iron-sulfur cluster and iron metabolism were up-regulated in S. intermedius (Fig. S9) as well as genes in several DNA-repair pathways including competence (uptake of extracellular DNA), RNA metabolism, and DNA modification and DNA repair enzymes.
3.4 Other
3.4.1 Oxygen had minor effects on expression of virulence genes
Despite the fact that the SMG is associated with anaerobic infections (e.g. abscesses), only a minority of genes associated with virulence are differentially regulated under oxygen varying growth conditions. Under anaerobic conditions, genes in oxidative stress pathway such as sialidase (nanA, 2.00-fold), pullulanase (pulA2, 2.17-fold), and a putative membrane toxin regulator (2.57-fold) were up-regulated (Fig. 3). nanA and pulA2 are associated with binding to host surfaces in streptococci (Hytönen et al. 2006; Brittan et al. 2012). On the other hand, some potential virulence genes such as proteases (Fig. 3), the bacteriocin accessory protein (bta, 4.00-fold) and a metallobetalactamase family protein (SIR_RS10820, 2.11-fold) were down-regulated anaerobically. The expression of CRISPR system (cas1, 2.53-fold; cas2, 2.01-fold; csn2, 2.70-fold) and nucleases (rnc, 2.01 fold; SIR_RS13205, 2.33 fold; hsdR, 2.93 fold) were increased anaerobically. These are involved in resistance to uptake of foreign genetic elements and phage infection (Marraffini and Sontheimer 2010; Midon et al. 2011; Sapranauskas et al. 2011; Derré-Bobillot et al. 2013). An association of the CRISPR system with oxidative stress tolerance has been previously seen in streptococci (Serbanescu et al. 2015). Overall, the expression of virulence associated genes was not strongly regulated by presence or absence of oxygen and these genes may be regulated by additional host specific signals.
3.4.2 Cellular membrane composition re-modelled to adapt to different environmental conditions
The major phospholipid (PL) class detected in S. intermedius was phosphatidyl glycerol (PG) and lyso-PG. There were 13 PGs and 2 lyso-PGs that varied significantly between aerobic and anaerobic growth conditions (Fig. 6). The cellular membrane of S. intermedius was largely composed of saturated short-chain PLs during aerobic growth. Consistent with the observed lipid profiles, the transcriptomic analysis found enoyl-CoA hydratase protein fabM (also referred to as phaB, 2.85-fold) and beta-ketoacyl-acyl carrier protein synthase III (fabH, 2.50-fold), associated with the synthesis of unsaturated and branched chain lipids respectively, were down-regulated under anaerobic conditions (Choi et al. 2000; Marrakchi et al. 2002; Fozo and Quivey 2004). It is unknown whether these metabolic and transcriptomic changes could affect the cellular membrane rigidity and permeability in S. intermedius. Increased levels of unsaturated fatty acids has been reported in E. faecalis during aerobic growth (Portela et al. 2014).

The heatmap of 15 statistically significantly affected phospholipids (p < 0.05) found in the intracellular metabolome of S. intermedius cultured in either aerobic or anaerobic conditions. The phospholipids were listed according to their alkyl chain length and saturation. The relative abundances of phospholipids were illustrated using a color scale, with blue indicating high abundances and red indicating low abundances (Color figure online)
4 Concluding remarks
This study examined the global physiologic, metabolic and transcriptomic adaptations of S. intermedius grown in aerobic and anaerobic environments. Our study demonstrates that while S. intermedius is able to adapt to either condition, the anaerobic growth condition is favored with a 24 % faster growth rate which also correlated with the up-regulation of the central carbon metabolism, the arginine deaminase pathway and the nucleotide de novo synthesis/salvage pathways. The largest transcriptional responses we observed were related to oxidative stress response under aerobic conditions. Overall, as a facultative anaerobe, S. intermedius is able to grow under varying oxygen tensions and may facilitate its colonization of distinct mucosal surfaces within the human host (upper respiratory, gastrointestinal and urogenital tracts). Moreover, its accelerated growth and adaptation to anaerobic conditions may reflect its propensity for polymicrobial pyogenic infections with anaerobic bacteria. This adaptability allows S. intermedius to coexist in complex polymicrobial environments, both as a commensal and a pathogen.
References
Ahn, S.-J., Ahn, S.-J., Browngardt, C. M., & Burne, R. A. (2009). Changes in biochemical and phenotypic properties of Streptococcus mutans during growth with aeration. Applied and Environment Microbiology, 75(8), 2517–2527.
Ahn, S.-J., Wen, Z. T., & Burne, R. A. (2007). Effects of oxygen on virulence traits of Streptococcus mutans. Journal of Bacteriology, 189(23), 8519–8527. doi:10.1128/JB.01180-07.
Asam, D., & Spellerberg, B. (2014). Molecular pathogenicity of Streptococcus anginosus. Mol Oral Microbiol, 29(4), 145–155.
Bassit, N., Boquien, C.-Y., Picque, D., & Georges, C. (1993). Effect of initial oxygen concentration on diacetyl and acetoin production by Lactococcus lactis subsp. lactis biovar diacetylactis. Applied and Environment Microbiology, 59(6), 1893–1897.
Brittan, J. L., Buckeridge, T. J., Finn, A., Kadioglu, A., & Jenkinson, H. F. (2012). Pneumococcal neuraminidase A: An essential upper airway colonization factor for Streptococcus pneumoniae. Molecular Oral Microbiology, 27(4), 270–283.
Broadhurst, D. I., & Kell, D. B. (2006). Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics, 2(4), 171–196.
Caspi, R., Altman, T., Billington, R., Dreher, K., Foerster, H., Fulcher, C. A., et al. (2014). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Research, 42(D1), D459–D471.
Choi, K.-H., Heath, R. J., & Rock, C. O. (2000). β-Ketoacyl-acyl carrier protein synthase III (FabH) is a determining factor in branched-chain fatty acid biosynthesis. Journal of Bacteriology, 182(2), 365–370.
Coman, G., Pânzaru, C., Diculencu, D., Gotia, D., Cârlan, M., Dahorea, C., & Butnaru, F. (1995). Pyogenic infections with different locations caused by Streptococcus anginosus alone or in association with anaerobic bacteria. Revista Medico-Chirurgicala A Societatii de Medici si Naturalisti din Iasi, 99(3–4), 215–219.
Cotter, P. D., & Hill, C. (2003). Surviving the acid test: Responses of gram-positive bacteria to low pH. Microbiology and Molecular Biology Reviews, 67(3), 429–453.
Creek, D. J., Dunn, W. B., Fiehn, O., Griffin, J. L., Hall, R. D., Lei, Z., et al. (2014). Metabolite identification: Are you sure? And how do your peers gauge your confidence? Metabolomics, 10(3), 350–353.
Crow, V. L., & Pritchard, G. G. (1977). Fructose 1,6-diphosphate-activated l-lactate dehydrogenase from Streptococcus lactis: Kinetic properties and factors affecting activation. Journal of Bacteriology, 131(1), 82–91.
Cusumano, Z. T., & Caparon, M. G. (2015). Citrulline protects Streptococcus pyogenes from acid stress using the arginine deiminase pathway and the F1F0-ATPase. Journal of Bacteriology, 197(7), 1288–1296.
Cusumano, Z. T., Watson, M. E., & Caparon, M. G. (2014). Streptococcus pyogenes arginine and citrulline catabolism promotes infection and modulates innate immunity. Infection and Immunity, 82(1), 233–242.
Derré-Bobillot, A., Cortes-Perez, N. G., Yamamoto, Y., Kharrat, P., Couvé, E., Da Cunha, V., et al. (2013). Nuclease A (Gbs0661), an extracellular nuclease of Streptococcus agalactiae, attacks the neutrophil extracellular traps and is needed for full virulence. Molecular Microbiology, 89(3), 518–531.
Fei, F., Bowdish, D. M. E., & McCarry, B. E. (2014). Comprehensive and simultaneous coverage of lipid and polar metabolites for endogenous cellular metabolomics using HILIC-TOF-MS. Analytical and Bioanalytical Chemistry, 406(15), 3723–3733.
Filkins, L. M., Hampton, T. H., Gifford, A. H., Gross, M. J., Hogan, D. A., Sogin, M. L., et al. (2012). Prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. Journal of Bacteriology, 194(17), 4709–4717.
Fozo, E. M., & Quivey, R. G. (2004). The fabM gene product of Streptococcus mutans is responsible for the synthesis of monounsaturated fatty acids and is necessary for survival at low pH. Journal of Bacteriology, 186(13), 4152–4158.
Gossling, J. (1988). Occurrence and pathogenicity of the Streptococcus milleri group. Reviews of Infectious Diseases, 10(2), 257–285.
Gruening, P., Fulde, M., Valentin-Weigand, P., & Goethe, R. (2006). Structure, regulation, and putative function of the arginine deiminase system of Streptococcus suis. Journal of Bacteriology, 188(2), 361–369.
Gupta, R., Yang, J., Dong, Y., Swiatlo, E., Zhang, J.-R., Metzger, D. W., & Bai, G. (2013). Deletion of arcD in Streptococcus pneumoniae D39 impairs its capsule and attenuates virulence. Infection and Immunity, 81(10), 3903–3911.
Higuchi, M. (1984). The effect of oxygen on the growth and mannitol fermentation of Streptococcus mutans. Journal of General Microbiology, 130(7), 1819–1826.
Higuchi, M., Yamamoto, Y., & Kamio, Y. (2000). Molecular biology of oxygen tolerance in lactic acid bacteria: Functions of NADH oxidases and Dpr in oxidative stress. Journal of Bioscience and Bioengineering, 90(5), 484–493.
Hirai, T., Kimura, S., & Mori, N. (2005). Head and neck infections caused by Streptococcus milleri group: An analysis of 17 cases. Auris, Nasus, Larynx, 32(1), 55–58.
Hocken, D. B., & Dussek, J. E. (1985). Streptococcus milleri as a cause of pleural empyema. Thorax, 40(8), 626–628.
Hytönen, J., Haataja, S., & Finne, J. (2006). Use of flow cytometry for the adhesion analysis of Streptococcus pyogenes mutant strains to epithelial cells: Investigation of the possible role of surface pullulanase and cysteine protease, and the transcriptional regulator Rgg. BMC Microbiology, 6(1), 18.
Imlay, J. A. (2013). The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nature Reviews Microbiology, 11(7), 443–454.
Jakubovics, N. S., Smith, A. W., & Jenkinson, H. F. (2002). Oxidative stress tolerance is manganese (Mn(2+)) regulated in Streptococcus gordonii. Microbiology, 148(Pt10), 3255–3263.
Kuhl, C., Tautenhahn, R., Böttcher, C., Larson, T.R., & Neumann, S. (2015). CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Analytical Chemistry, 84(1), 283–289.
Laupland, K. B., Ross, T., Church, D. L., & Gregson, D. B. (2006). Population-based surveillance of invasive pyogenic streptococcal infection in a large Canadian region. Clinical Microbiology & Infection, 12(3), 224–230.
Lee, E. Y., Khatwa, U., McAdam, A. J., Bastos, M. Almeida, Mahmood, S. A., Ervoes, J. P., & Boiselle, P. M. (2010). Streptococcus milleri group pleuropulmonary infection in children: Computed tomographic findings and clinical features. Journal of Computer Assisted Tomography, 34(6), 927–932.
Marquis, R. E., Bender, G. R., Murray, D. R., & Wong, A. (1987). Arginine deiminase system and bacterial adaptation to acid environments. Applied and Environment Microbiology, 53(1), 198–200.
Marraffini, L. A., & Sontheimer, E. J. (2010). CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nature Reviews Genetics, 11(3), 181–190.
Marrakchi, H., Zhang, Y.-M., & Rock, C. O. (2002). Mechanistic diversity and regulation of Type II fatty acid synthesis. Biochemical Society Transactions, 30(part 6), 1050–1055.
Midon, M., Schäfer, P., Pingoud, A., Ghosh, M., Moon, A. F., Cuneo, M. J., et al. (2011). Mutational and biochemical analysis of the DNA-entry nuclease EndA from Streptococcus pneumoniae. Nucleic Acids Research, 39(2), 623–634.
Olson, A. B., Kent, H., Sibley, C. D., Grinwis, M. E., Mabon, P., Ouellette, C., et al. (2013). Phylogenetic relationship and virulence inference of Streptococcus anginosus group : Curated annotation and whole-genome comparative analysis support distinct species designation. BMC Genomics, 14, 895.
Parkhomchuk, D., Borodina, T., Amstislavskiy, V., Banaru, M., Hallen, L., Krobitsch, S., et al. (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Research, 37(18), e123.
Parkins, M. D., Sibley, C. D., Surette, M. G., & Rabin, H. R. (2008). The Streptococcus milleri group—an unrecognized cause of disease in cystic fibrosis: A case series and literature review. Pediatric Pulmonology, 43(5), 490–497.
Portela, C. A. F., Smart, K. F., Tumanov, S., Cook, G. M., & Villas-Bôas, S. G. (2014). Global metabolic response of Enterococcus faecalis to oxygen. Journal of Bacteriology, 196(11), 2012–2022.
Prüb, B. M., Nelms, J. M., Park, C., & Wolfe, A. J. (1994). Mutations in NADH: Ubiquinone oxidoreductase. Journal of Bacteriology, 176(8), 2143–2150.
Ripley, R. T., Cothren, C. C., Moore, E. E., Long, J., Johnson, J. L., & Haenel, J. B. (2006). Streptococcus milleri infections of the pleural space: Operative management predominates. American Journal of Surgery, 192(6), 817–821.
Rogers, A. H., Zilm, P. S., & Gully, N. J. (1987). Influence of arginine on the coexistence of Streptococcus mutans and S. milleri in glucose-limited mixed continuous culture. Microbial Ecology, 14(3), 193–202.
Ruoff, K. L. (1988). Streptococcus anginosus (“Streptococcus milleri”): the unrecognized pathogen. Clinical Microbiology Reviews, 1(1), 102–108.
Sapranauskas, R., Gasiunas, G., Fremaux, C., Barrangou, R., Horvath, P., & Siksnys, V. (2011). The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Research, 39(21), 9275–9282.
Serbanescu, M. A., Cordova, M., Krastel, K., Flick, R., Beloglazova, N., Latos, A., et al. (2015). Role of the Streptococcus mutans CRISPR-Cas systems in immunity and cell physiology. Journal of Bacteriology, 197(4), 749–761.
Sezonov, G., Joseleau-Petit, D., & D’Ari, R. (2007). Escherichia coli physiology in Luria-Bertani broth. Journal of Bacteriology, 189(23), 8746–8749.
Shinzato, T., & Saito, A. (1994). A mechanism of pathogenicity of “Streptococcus milleri group” in pulmonary infection: Synergy with an anaerobe. Journal of Medical Microbiology, 40, 118–123.
Shinzato, T., & Saito, A. (1995). The Streptococcus milleri group as a cause of pulmonary infections. Clinical Infectious Diseases, 21(Supplement 3), S238–S243.
Sibley, C. D., Church, D. L., Surette, M. G., Dowd, S. E., & Parkins, M. D. (2012). Pyrosequencing reveals the complex polymicrobial nature of invasive pyogenic infections: Microbial constituents of empyema, liver abscess, and intracerebral abscess. European Journal of Clinical Microbiology and Infectious Diseases, 31(10), 2679–2691.
Sibley, C. D., Parkins, M. D., Rabin, H. R., Duan, K., Norgaard, J. C., & Surette, M. G. (2008). A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America, 105(39), 15070–15075.
Siegman-Igra, Y., Azmon, Y., & Schwartz, D. (2012). Milleri group streptococcus—a stepchild in the viridans family. European Journal of Clinical Microbiology and Infectious Diseases, 31(9), 2453–2459.
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R., & Siuzdak, G. (2006). XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Analytical Chemistry, 78(3), 779–787.
Stadelmann, B., Hanevik, K., Andersson, M. K., Bruserud, O., & Svärd, S. G. (2013). The role of arginine and arginine-metabolizing enzymes during Giardia—host cell interactions in vitro. BMC Microbiology, 13, 256.
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis chemical analysis working group (CAWG) metabolomics standards initiative (MSI). Metabolomics, 3(3), 211–221.
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3), 562–578.
Trotter, E. W., Rolfe, M. D., Hounslow, A. M., Craven, C. J., Williamson, M. P., Sanguinetti, G., et al. (2011). Reprogramming of Escherichia coli K-12 metabolism during the initial phase of transition from an anaerobic to a micro-aerobic environment. PLoS One, 6(9), e25501.
Van der Auwera, P. (1985). Clinical significance of Streptococcus milleri. European Journal of Clinical Microbiology, 4(4), 386–390.
Whiley, R. A. A., Beighton, D., Winstanley, T. G. G., Fraser, H. Y. Y., & Hardie, J. M. M. (1992). Streptococcus intermedius, Streptococcus constellatus, and Streptococcus anginosus (the Streptococcus milleri group): association with different body sites and clinical infections. Journal of Clinical Microbiology, 30(1), 243–244.
Wong, C. A., Donald, F., & Macfarlane, J. T. (1995). Streptococcus milleri pulmonary disease: A review and clinical description of 25 patients. Thorax, 50(10), 1093–1096.
Xia, J., Sinelnikov, I. V., Han, B., & Wishart, D. S. (2015). MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Research, 43(W1), W251–W257.
Zheng, L., T’Kind, R., Decuypere, S., von Freyend, S. J., Coombs, G. H., & Watson, D. G. (2010). Profiling of lipids in Leishmania donovani using hydrophilic interaction chromatography in combination with Fourier transform mass spectrometry. Rapid Communications in Mass Spectrometry, 24, 2074–2082.
Zúñiga, M., Pérez, G., & González-Candelas, F. (2002). Evolution of arginine deiminase (ADI) pathway genes. Molecular Phylogenetics and Evolution, 25(3), 429–444.
Acknowledgments
This work is dedicated to the late Prof. Brian McCarry (1946–2013). FF was supported by an Ontario Graduate Scholarship. MGS and DMEB are supported by the CIHR and hold Canada Research Chairs. Work in the Bowdish laboratory is supported by the McMaster Immunology Research Centre (MIRC) and work in the Bowdish and Surette labs are supported by the M.G. DeGroote Institute for Infectious Disease Research (IIDR). The authors would like to thank the Center for Microbial Chemical Biology (CMCB) at McMaster for access to the LC–MS. This study was funded by a grant from the Canadian Institutes of Health Research to DMEB and MGS (Grant # 108032).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declared no conflict of interest.
Human and animal informed consent
This article does not contain any studies with human participants or animals performed by any of the author.
Additional information
Fan Fei, and Michelle L. Mendonca have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fei, F., Mendonca, M.L., McCarry, B.E. et al. Metabolic and transcriptomic profiling of Streptococcus intermedius during aerobic and anaerobic growth. Metabolomics 12, 46 (2016). https://doi.org/10.1007/s11306-016-0966-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-016-0966-0