
Abstract
The polymicrobial nature of invasive pyogenic infections may be underestimated by routine culture practices, due to the fastidious nature of many organisms and the loss of viability during transport or from prior antibacterials. Pyrosequencing was performed on brain and liver abscesses and pleural fluid and compared to routine culture data. Forty-seven invasive pyogenic infection samples from 44 patients [6 intracerebral abscess (ICA), 21 pyogenic liver abscess (PLA), and 18 pleural fluid (PF) samples] were assayed. Pyrosequencing identified an etiologic microorganism in 100 % of samples versus 45 % by culture, p <0.01. Pyrosequencing was also more likely than traditional cultures to classify infections as polymicrobial, 91 % versus 17 %, p <0.001. The median number of genera identified by pyrosequencing compared to culture was 1 [interquartile range (IQR) 1–3] versus 0 (IQR 0–1) for ICA, 7 (IQR 1–15) versus 1 (IQR 0–1) for PLA, and 15 (IQR 9–19) versus 0 (IQR 0–1) for PF. Where organisms were cultured, they typically represented the numerically dominant species identified by pyrosequencing. Complex microbial communities are involved in invasive pyogenic infection of the lung, liver, and brain. Defining the polymicrobial nature of invasive pyogenic infections is the first step towards appreciating the clinical and diagnostic implications of these complex communities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Invasive pyogenic infections are responsible for a large burden of disease in general populations [1]. Local tissue invasion by pathogenic bacteria and the resultant formation of a well-vascularized capsule and containment of infection through a focused host immune response characterize pyogenic infections. Abscesses can occur at any sterile site within the human body; however, the pathophysiologic mechanisms involved in each have unique features. The microbial pathogens classically associated with abscess formation have been derived from decades of traditional culture-based microbial practices and are relatively conserved between different abscess types. While the vast majority of invasive infections yield only a single pathogen (monomicrobial), polymicrobial infections have been demonstrated to occur in at least 11–18 % of intracerebral abscesses (ICAs) [2, 3], 11–49 % of pyogenic liver abscesses (PLAs) [4–6], and 10–15 % of empyemas [7]. This value is thought to under-represent the true diversity of polymicrobial infections owing to the limitations of traditional culture techniques, effects of prior antibacterials, loss of organism viability during transport, and the presence of fastidious or uncultivable microorganisms. Furthermore, our understanding of bacterial infectious diseases is changing through the application of culture-independent technologies to complement traditional culture-based techniques. Recent work has shown that the diversity of bacterial species is much greater than originally thought in models of surgical site infections [8], diabetic foot infections [9, 10], brain abscesses [11, 12], and cystic fibrosis lower airways disease [13–16].
We hypothesized that traditional culture-based recovery of organisms present in invasive pyogenic collections likely underestimates the abundance and diversity of organisms present in abscesses. We sought to compare the microbial constituents identified in ICA, PLA, and empyema specimens by using culture-dependent and culture-independent approaches (pyrosequencing). The laboratory results were then used to determine if an expanded spectrum of causative microbial pathogens influenced patient outcomes.
Methods
Study population
Randomly selected samples of patients with suspected pyogenic infections during the period July 2009–October 2010 were identified from a centralized laboratory servicing four acute care hospitals within the Calgary Health Zone (CHZ). Clinical, demographic, microbiological, treatment, and outcome information for these patients was collected through a detailed chart review and diagnosis was confirmed. Approval was obtained from the Conjoint Health Research Ethics Board at the University of Calgary and the CHZ prior to commencement of this study.
Pyogenic samples
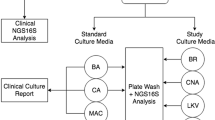
Clinical specimens were cultured aerobically and anaerobically. Aerobic culture was performed on blood agar, MacConkey agar, and colistin–nalidixic acid media at 35 °C in 5 % CO2 for 48 h. Anaerobic culture was performed using brucella, kanamycin–vancomycin laked blood (KVLB), Bacteroides bile esculin (BBE), and phenylethyl alcohol (PEA) agar in the Anoxomat (Mart Microbiology, AJ Drachten, the Netherlands) anaerobic system environment at 35 °C for 96 h. Isolates were identified to the species level using standard biochemical methods using the automated Vitek 2 system (bioMérieux, Laval, Quebec, Canada), and partial sequencing of the 16S rRNA gene according to standard methods [17, 18]. Antibiotic susceptibility testing was performed on all isolates according to published guidelines [17, 19]. The remaining portion of each pyogenic sample was stored at −80 °C for an extended period of time and then analyzed en masse to determine their molecular constituents.
DNA extraction
Pus/tissue was homogenized and 200 mg was aseptically suspended in 500 μl RLT buffer with β-mercaptoethanol (Qiagen, Valencia, CA). A sterile 5-mm steel bead (Qiagen, Valencia, CA) and 500-μl volume of sterile 0.1-mm glass beads (Scientific Industries, Inc., NY, USA) were used for complete bacterial lyses in a Qiagen TissueLyser (Qiagen, Valencia, CA) at 30 Hz for 5 min. Samples were centrifuged, and 100 μl of 100 % ethanol was added to a 100-μl aliquot of the sample supernatant. This mixture was added to a DNA spin column and DNA extraction protocols were followed (Qiagen, Valencia, CA). DNA was eluted from the column with 50 μl of sterile water and samples were diluted to a final concentration of 20 ng/μl. DNA samples were quantified using a NanoDrop spectrophotometer (Nyxor Biotech, Paris, France).
Massively parallel bTEFAP
Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) was performed as previously described using Gray28F 5′-TTTGATCNTGGCTCAG and Gray519r 5′-GTNTTACNGCGGCKGCTG [9, 10, 16, 20, 21]. Tag-encoded FLX amplicon pyrosequencing analyses utilized a Roche 454 FLX instrument with Titanium series reagents and procedures.
Bacterial diversity analysis
Following sequencing, all failed sequence reads, products less than 350 bp in length, and reads of low-quality sequences were removed; tags and primers were removed. Sequence collections were depleted of any non-bacterial ribosome sequences and chimeras using B2C2 [22], as previously described [9, 10, 16, 20, 21]. To determine the identity of bacteria in the sequence collection, sequences were denoised, assembled into clusters, and queried with a distributed BLASTn .NET algorithm [23] against a database of high-quality bacterial 16S rRNA sequences from the NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Database sequences were characterized as high quality based upon similar criteria utilized by the Ribosomal Database Project (RDP) version 9 [24]. Using a .NET and C# analysis pipeline, the resulting BLASTn outputs were compiled, validated using taxonomic distance methods, and data reduction analysis was performed as described previously [9, 10, 16, 20, 21]. Inclusion criteria for further analysis included greater than 1,000 sequence reads per specimen.
Taxonomic determination
Based upon the BLASTn-derived sequence identity (percentage of the total length of the query sequence which aligns with a given database sequence) that was validated using taxonomic distance methods, bacteria were classified at the appropriate taxonomic levels based upon the following criteria. Sequences with identity scores to known or well characterized 16S rRNA sequences greater than 95 % were resolved to the genus level, 90–95 % to the family level, 85–90 % to the order level, 80–85 % to the class level, and 77–80 % to the level of phyla. After resolving based upon these parameters, the percentage of each bacterial ID was individually analyzed for each sample, providing relative abundance information within and among the individual samples. Evaluations presented at each taxonomic level, including percentage compilations, represent all sequences resolved to their primary identification or their closest relative, as has been previously described [16]. Organisms identified with sequence reads representing less than 1 % of the total population (i.e., <10 sequence reads per 1,000 total sequence reads) were excluded from the analysis.
Definitions
Abscesses were subcategorized according to their pathogenesis as identified by a detailed chart review [25]. ICAs were defined as being either odontogenic, sinus, otitis, penetrating/nosocomial, or hematogenous seeding in origin. PLAs were defined as being biliary, hepatic artery (i.e., hematogenous seeding), portal vein (intraabdominal/visceral/bowel in origin), direct extension from contiguous focus of infection, penetrating trauma, or amebic liver abscess in origin. Exudative effusions were classified as complex parapneumonic effusions or empyemas [26]. Bacteremia was defined by the isolation of an organism from one or more sets of aseptically obtained blood culture bottles [27].
Prior antibacterial therapy was classified based on the intended spectrum of activity against abscess pathogens [25]. Regimens containing metronidazole, clindamycin, carbapenem, or a β-lactam/β-lactamase inhibitor combination were classified as having “anaerobic activity”. Regimens including a fluoroquinolone, macrolide, or tetracycline were described as having “atypical activity”. Regimens containing either a fluoroquinolone, aminoglycoside, a second-, third-, or fourth-generation cephalosporin, carbapenem, or β-lactam/β-lactamase inhibitor combination were classified as having “Gram-negative activity”. Vancomycin was analyzed separately from other Gram-positive active drugs. “Gram-positive active” drugs were considered to be first-generation cephalosporins, ceftriaxone/cefotaxime, fourth-generation cephalosporins, carbapenems, β-lactam/β-lactamase inhibitor combinations, or a third- or fourth-generation fluoroquinolone.
In an analysis focused on bacterial DNA, antibiotic susceptibility testing was obviously not possible. For the purposes of this assessment, several assumptions were made regarding the susceptibility of organisms based on the genus-level identification [25, 28–30]. Enterococcus spp. were assumed to be resistant to all cephalosporin antibiotics. Stenotrophomonas spp. were assumed to be resistant to all cephalosporins and carbapenems. Pseudomonas spp. were assumed to be resistant to aminopenicillins, first- and second-generation cephalosporins, and ceftriaxone/cefotaxime. The tribe Proteeae were assumed to have resistance to first-, second-, and third-generation cephalosporins. Organisms that were obligate anaerobes were presumed to be resistant to first-generation cephalosporins. Atypical organisms (such as Mycoplasma and Chlamydia) were assumed to be resistant to cell wall-active agents.
Nosocomial-associated (NA) infections were those occurring ≥48 h after hospital admission or within 48 h of discharge from the hospital. Community-associated (CA) infections were those which were first identified <48 h after hospital admission or >48 h after discharge. Infections were defined as being healthcare-associated, community-associated (HCA-CA) if the patients had recent prior contact with the healthcare system, as previously described [27]. CA infections were those which were of community onset that were not healthcare associated.
Statistical analysis
Individual variables were assessed using histograms prior to analysis in order to identify their underlying distribution. Variables with normal or near-normal distributions were described with means and standard deviations (SDs) and compared using Student’s t-test. Medians with interquartile ranges (IQRs) were used to describe non-normally distributed variables and compared using the Mann–Whitney U-test. Categorical variables were compared using Fisher’s exact test. All statistical analyses were performed using Stata version 11.0 (Stata Corp., College Station, TX, USA).
Results
Patient characteristics
Over 15 months, 102 randomly selected clinical samples submitted for aerobic/anaerobic culture from 94 patients with presumptive invasive pyogenic infections were identified and immediately frozen at −80 °C. All of the study samples analyzed had been collected as part of each patient’s routine care. After detailed clinical review, only 57 samples from 52 patients collected over 12 months were confirmed to represent invasive pyogenic infection (other samples that were excluded included cancerous lesions or uninfected cysts, etc). Of these, 47 samples from 44 patients met the a priori criteria of yielding >1,000 sequence reads and were included in the final analysis.
Eight brain abscess samples were procured from six patients. The median age of the patients with ICAs was 36 years (IQR 4–50). Etiologies were odontogenic in four [right frontal (2), right fronto-temporal (1), left fronto-temporal (1)], sinus in one (right frontal), and hematogenous in one (thalamic). The co-morbidities of patients with ICAs are listed in Table 1. Three cases were CA and three were HCA-CA. Recognized risk factors for the acquisition of ICA included alcoholism in 3/6 cases and active chemotherapy for breast (1/6) and prostate cancer (1/6 cases).
Twenty-one liver abscess samples were procured from 20 patients. The median age of the patients was 61 years (IQR 51–72). Five cases were NA, three cases HCA-CA, and 12 cases were CA. Etiologies were infected cyst (1), biliary 4 (right lobe 3, left lobe 1), portal vein 13 (11 right lobe, 2 multiple focus), and one each of direct extension from a contiguous source of infection (right lobe) and hepatic artery (right lobe). Identified risk factors included chronic biliary disease 2/20 (10 %), alcoholism or substance abuse 4/20 (20 %), diabetes mellitus 4/20 (20 %), cancer 4/20 (20 %), and inflammatory bowel disease 3/20 (15 %).
Eighteen pleural samples were procured from 18 patients; five had complex parapneumonic effusions (four right-sided, one left-sided), and 13 had empyemas (nine left-sided, four right-sided). Seven cases were NA, four cases were HCA-CA, and seven cases were CA. Identified co-morbidities for those with empyemas included chronic respiratory disease 9/18 (50 %), diabetes mellitus 7/18 (30 %), cancer 4/18 (22 %), and alcoholism and/or substance abuse 4/18 (22 %).
Microbial constituents
Pyrosequencing yielded 269,303 total sequence reads from the 47 included pyogenic samples. The median number of sequence reads for ICAs was 7,540 (IQR 4,839–11,747), liver 6,555 (IQR 3,292–10,151), and lung 2,667 (IQR 1,364–6,336).
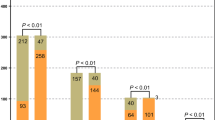
Routine culture practices were less likely to identify an etiologic microorganism [45 % of samples (21/47)] as compared to pyrosequencing [100 % of samples (47/47)], p <0.001. Pyrosequencing was more likely than traditional cultures to identify polymicrobial infections [91 % (43/47) vs. 17 % (8/47)], p <0.001.
Pyrosequencing revealed that the majority of bacterial genera recovered from the brain were Gram-positive organisms, with obligate anaerobes being uncommon components of ICAs (Fig. 1). Within ICAs, four samples yielded an etiological organism by culture, all growing monomicrobial Streptococcus milleri group (SMG) as the sole pathogen. Pyrosequencing confirmed three of these to be monomicrobial with organisms within the genus Streptococcus, accounting for >99.9 % of the total sequence reads. However, the fourth revealed the Streptococcus genus to account for 38.1 % of sequence reads, with significant concentrations of Fusobacterium spp. (53.2 %) and Clostridium spp. (8.0 %). Four samples failed to yield an organism through conventional culture-based approaches but had etiologic agents identified as a result of prior microbiologic testing [SMG bacteremia (2), and CONS and Propionibacterium spp. from abscess specimens from the same infection at a different point in time (2)]. Culture-independent analysis indicated that three of these specimens were polymicrobial (2, 7, and 27 genera identified) and one was monomicrobial. In those ICA samples with an organism identified by culture, the cultured organism accounted for the majority of sequences identified (97 %; range 38–100 %) (Fig. 2).

Microbial community composition of pyogenic specimens. a Classified as either predicted Gram-positive or Gram-negative bacteria. b Community members as determined by the presence of predicted obligate anaerobes (each individual member having to be present at a rate of >1 % of the total population)

Identified constituent organisms of pyogenic specimens as a function of relative abundance within the community, as measured by pyrosequencing. a Intracerebral brain abscess. b Pyogenic liver abscess. c Pulmonary empyema/complicated pleural effusion. Those organisms identified molecularly that were also recovered using culture-based approaches are illustrated within the accompanying legend
Eight PLA samples were culture-negative, 11 samples were monomicrobial (E. coli 4, Proteus mirabilis 3, and one each of Candida albicans, Bacillus spp., SMG Fusobacterium spp., and Propionibacterium acnes), two samples were polymicrobial [one with E. coli, S. aureus, SMG, and Streptococcus viridans group (SVG), and the other growing Klebsiella pneumonia, Citrobacter koseri, and SMG]. Pyrosequencing demonstrated a much broader range of organisms; a median of seven genera per sample (IQR 1–15, range 1–22) versus culture, which yielded one genus per samepl (IQR 0–1, range 0–4). The majority of genera in PLA samples were Gram-positives (Figs. 1 and 3). The most commonly identified organisms from PLAs (identified at concentrations greater than 5 % in at least two samples) included: Streptococcus, Lactobacillus, Stenotrophomonas, Enterobacter, Escherichia, and Clostridium (Fig. 4). Culture failed to identify low-abundance organisms and anaerobes/nutritionally fastidious organisms not previously described in PLAs (Fig. 2). In PLAs where an organism(s) was identified by culture, it accounted for a median of 86 % of sequence reads (IQR 13–89 %; range 1–99 %), indicating that culture-based approaches are effective at recovering numerically dominant organisms. Anaerobes were found in 71 % of samples (Fig. 1) and often comprised the dominant community members (Figs. 3 and 4).

Relative frequency of microorganisms identified in pyogenic infections. Only organisms which account for ≥5 % of sequences within an individual sample are included for ease of visualization. The relative distance between each individual black line and the total abundance of each organism indicates the proportion of samples in which this organism was a numerically dominant member of the community

Microbial composition of individual abscesses. Only organisms that account for ≥5 % of the total number of sequences within an individual sample are included for ease of visualization. The solid black lines around each respective color identify Gram-positive aerobes. Gram-positive obligate anaerobes are identifiable by striped vertical bars. The dashed black lines around each respective color indicate Gram-negative aerobes. The angled bars indicate Gram-negative anaerobes. Parapneumonic effusion are indicated by an X, OF denotes oropharyngeal flora, and E represents (classically) environmental isolates. *Several genera are grouped; Gram-negative environmental anaerobic organisms included: Leptotrichia, Tannerella, Merismopedia, Levilinea, and Roseobacter. Gram-negative environmental aerobic organisms included: Marinobacterium, Acidobacteriaceae, Hyphomonadaceae, Comamonadaceae, Pelagibacter, Rubellimicrobium, Agarivorans, Methylophilales, Nautella, Acidithiobacillus, Sphingobacterium, and Sulfitobacter. Gram-positive anaerobic organisms included: Peptoniphilus, Lachnospiraceae, Oscillospira, Ferrovum, Anaerococcus, Micrococcineae, and Clostridiisalibacter. Gram-positive aerobic organisms included: Janibacter, Conexibacter, and Arthromitus. The tribe Proteeae includes Enterobacter, Citrobacter, and Proteus
In 18 empyema/parapneumonic effusion cultures, there were three culture-positive samples; one monomicrobial (CONS) and two polymicrobial (methicillin-resistant Staphylococcus aureus and CONS, and S. pneumoniae and Peptostreptococcus spp.). These organisms accounted for a median of 64 % (IQR 27–80 %) of sequences identified from those culture-positive samples. Considerably greater diversity was identified in microbial constituents by using a culture-independent approach; a median of 15 genera by pyrosequencing (IQR 9–19, range 3–27) versus a median of zero genera by culture (IQR 0–0, range 0–2). Specimens from the pleural cavity were the only sample type where it was more common to detect Gram-negatives than Gram-positives (Fig. 1). The vast majority (89 %) of these samples contained obligate anaerobes, often as the dominant community member (Figs. 3 and 4).
Bacteremia was more likely to accompany specific infections; 45 % (9/20) PLA, 17 % (1/6) ICA cases, and 17 % (3/18) respiratory pleural infections, p = 0.05. Bacteremias were monomicrobial in 91 % (10/11) of episodes and polymicrobial in 9 % (1/11). The most frequent bacteremic isolates were: SMG (1 ICA, 3 PLA), followed by SVG (2 PLA), E. coli (2 PLA), and one of each C. perfringens (PLA), Corynebacterium amycolatum (empyema), methicillin-resistant Staphylococcus aureus (empyema), Enterococcus faecium (PLA), and Enterobacter cloacae (PLA). In bacteremic patients, the organism recovered by the blood culture was also the most abundant organism identified by pyrosequencing in 91 % (10/11) of episodes, accounting for a median of 78 % of the total sequence reads (IQR 39–97 %).
Taxonomic classification at the family level revealed that 10 % (2/20), 16.3 % (7/43), and 38.1 % (24/64) of the organisms recovered from brain, liver, and lung samples were unique to that specific specimen type, respectively (Fig. 5). A detailed list of these families is provided in Table 2. Eleven bacterial families were detected in all pyogenic specimens (from at least two samples).

Venn diagram displaying abscess microbial constituents organized into bacterial families. To be included, each organism had to be identified in >1 % of at least two independent samples. The superscripts indicate the number of order-level-only classifications within each category. A detailed breakdown is available in Table 2
Effects of prior empiric antibiotics
Empiric antibacterial therapies were provided before culturing in 44/47 samples (94 %). The median duration of antibiotic exposure before sampling was 6 days (IQR 1.2–11 days; range 0–53 days). Antibiotic regimens administered to the six patients with ICAs before aspiration had broad-spectrum Gram-positive [83 % (5/6)] and Gram-negative activity [83 % (5/6)], and were supplemented with vancomycin [83 % (5/6)] and occurred for a median of 4 days (IQR 0.1–53 days). Anaerobic treatment was provided in 83 % (5/6). No ICAs were treated with antibiotics with atypical organism coverage. Empiric regimens provided before the sampling of PLAs in 20 patients contained broad-spectrum Gram-positive [75 % (15/20)] and Gram-negative activity [95 % (19/20)], and were supplemented with vancomycin [15 % (3/20)] and occurred for a median of 8 days (IQR 1–12 days) before aspiration. These regimens covered for anaerobic microorganisms or atypical organisms in 90 % (18/20) and 30 % (6/20) of cases, respectively. Empyema cases were treated for a median of 5 days (IQR 2.2–9.5 days) before thoracentesis and contained Gram-positive [78 % (14/18)] and Gram-negative [89 % (16/18)] activity supplemented with vancomycin in 33 % (6/18) of cases. The regimens had anaerobic and atypical activity in 61 % (11/18) and 78 % (14/18) of cases, respectively.
The number of organisms recovered from culture-based approaches decreased with antibiotic exposures beyond one day; median one versus zero, p = 0.001. However, bacterial nucleic acid could be identified in all samples. As expected, the number of bacterial organisms detected by culture-independent processes did not change as a result of empiric therapy stratified by more or less than 1 day (mean 10.4 vs. 10.2 genera/sample, p = 0.91), or more or less than 5 days (median 9.6 vs. 9.2 genera/sample, p = 0.61). The emergence of bacteria intrinsically resistant to antibacterial agents was not observed with a longer duration of empiric antibacterial therapies when comparing the results of samples taken before or after 1 and 5 days of therapy (Table 3).
Treatment and outcomes
All patients were treated for invasive pyogenic infections. The median treatment duration for ICAs was 66 days (IQR 61–71 days), 50 days for PLAs (IQR 34–81 days), and 44.5 days (IQR 25–49 days) for empyema. Intensive care unit support was required for 3/6 (50 %) cases of ICA, 1/20 (5 %) cases of PLA, and 9/18 (50 %) cases of empyema. All patients initially received therapy with more than one antibacterial agent. Only 11 % (5/44) of the patients had their initial empiric regimen narrowed to account for only those organisms identified in culture.
The all-cause in-hospital mortality was 0/6 (0 %) for ICAs, 2/20 (10 %) for PLAs, and 3/18 (17 %) for empyemas, p = 0.55. Patients who died were no more likely to have positive abscess clinical cultures: 60 % (3/5) versus 44 % (17/39), p = 0.64. Pyogenic samples with organisms presumed to be intrinsically resistant to specific classes of antibacterials were no more likely to require a prolonged course of antibiotics and did not have an increased risk of death (Table 4). In those patients whose treatment regimen lacked adequate antimicrobial activity against intrinsically resistant abscess component flora, there were no differences noted in either their treatment duration or outcome compared to those on appropriate therapy (Table 5).
Discussion
Several groups have explored the use of pyrosequencing to determine the richness of bacterial communities of various human sites. Whereas studies of the microbiome of the gastrointestinal tract [31, 32], superficial wounds [8, 10, 33–35], and lower airways of patients with chronic respiratory disease [14, 16, 36] must contend with the issue of external environmental contamination of samples, pyogenic infections represent invasion into normally sterile sites. As such, it can be assumed that any bacterial DNA present within an abscess is foreign in nature.
A comprehensive understanding of the microbial constituents is fundamental to understanding the pathophysiology of pyogenic infections. Communication between bacterial community members have been shown to alter the virulence factor production of pathogens [37]. Therefore, the microbial community context in which pathogens exist during infection may be important in understanding clinical outcomes. In animal models of infection, even presumed benign commensal organisms have the ability to increase the pathogenicity of traditionally accepted pathogens [38, 39]. Community diversity may enable the development of a microniche, whereby different community members modify local environments to be more or less favorable to other community members [40, 41]. The impairment of host immune response may similarly be afforded through intraspecies phagocytic inhibition [40]. Finally, minor community members may have the ability to limit the effects of clinical intervention through the production of secreted antibiotic-modifying enzymes which may serve to protect the entire community [42–44].
The data herein have demonstrated that the distribution of organisms involved in pyogenic infection to be more diverse than that recovered through routine clinical culture practice. Indeed, polymicrobial infection is the norm, as opposed to the exception in PLAs and empyemas. SMG was the most commonly cultivated pathogen in brain and liver abscess samples. Indeed, this organism is often reported as the most common organism from a variety of pyogenic infections, including: brain abscess [45–47], liver abscess [48, 49], and empyema [50, 51]. We found streptococcal DNA to be present in high abundance in all specimen types.
Only one other group has explored pyrosequencing to examine the polymicrobial community structure of pyogenic samples [11, 12]. In their initial study, Al Masalma et al. compared culture to pyrosequencing in patients with ICAs and found that an etiologic agent could be identified in all cases, that one-quarter of the presumed monomicrobial infections were indeed polymicrobial, and that the diversity of organisms in polymicrobial specimens was under-represented by culture [11]. Our results are in accord with these findings. In their most recent publication which included a much larger number of patient samples, they were even able to identify that the etiology of ICAs correlated with the microbiologic constituents (otitis media-associated cases were often monomicrobial, whereas dental- and sinusitis-associated cases were polymicrobial) [12].
The most frequently cultured organisms from PLAs in published series include Enterobacteriaceae, such as E. coli and Klebsiella spp., and SMG [5, 6, 52]. These genera were commonly identified in our pyrosequencing studies and, furthermore, represented the numerically abundant organisms in those samples that were culture-positive. However, a much greater diversity of minor community members was observed using pyrosequencing, with a seven-fold increase in the number of community members identified relative to traditional culture. Most notably, anaerobes were identified in more than three-quarters of samples as compared to published series, where these are found in less than 20 % of samples [5, 53, 54].
Most intriguingly was the information from the pleural infection samples. Whereas organisms are recovered from traditional culture in less than 15 % of empyema samples [50, 51], pyrosequencing detected bacteria in 100 % of samples. Furthermore, the greatest microbial diversity from each of the pyogenic infections was found in pleural samples. Anaerobic microorganisms were found to predominate pleural samples in both the number of genera and the total relative abundance, with many of these anaerobes being common members of the oropharynx [55]. A great number of organisms classically described only in marine and freshwater environments were also identified and likely represent minor components of the oropharynx [55, 56]. Within normal healthy hosts, it has been demonstrated that the respiratory microbiome constituents are highly distinct from individual to individual [56, 57] and, as such, partially explains the diversity in the pleural samples assessed. Finally, no difference between complicated parapneumonic effusion samples and those of empyemas were observed, suggesting that these processes exist along a continuum.
In order to improve patient outcomes, empiric broad-spectrum anti-bacterial therapy is recommended for the management of deep-tissue pyogenic infection once infection is suspected, regardless of the microbiologic sampling. Prior therapy is associated with lower rates of pathogen identification by culture [58, 59]. Remarkably, pyrosequencing can identify bacterial DNA from pyogenic collections, even after more than 7 weeks of antibacterial therapy.
Many of the infections from our cohort were associated with minor component members that were inherently resistant to the antibacterial agents to which they were treated. However, this cross-sectional data suggests that infections with organisms with intrinsically resistant organisms were no more likely to result in protracted therapy or increase the risk of death even when these therapies were not provided. This is in accord with the clinical data from a number of situations, including diabetic foot infections [60, 61] and intraabdominal abscess [62]. Notably, in intraabdominal infections, despite their association with worse clinical outcomes, therapies with anti-enterococcal coverage do not result in improved outcomes relative to those that are enterococcal-sparing [62–64]. However, clinicians treat invasive pyogenic infections as polymicrobial, with broad-spectrum antibiotics for a pre-specified time periods, often without narrowing their regimen based on culture results, acknowledging the inherent limitations of culture results. As such, retrospective studies evaluating treatment duration may be too insensitive to allow assessing the influence of the polymicrobial community structure on clinical outcomes. It is possible that pyrosequencing may provide clinicians the confidence to offer narrow-spectrum antibiotics for shorter periods of time, thus, reducing antibiotic consumption and reducing the risk of emerging resistance. Prospective comparative studies evaluating pyrosequencing-supplemented culture results to the standard of care are required.
It is unlikely that all microorganisms identified within an abscess must be specifically targeted with antibacterial therapies. What is not clear, however, is which microorganisms do require targeted therapy. Is it sufficient to merely target the dominant community members—those identified by routine anaerobic culture practice? Could focused therapy against a particular minor but crucial community member be sufficient to enable cure? Given the potential of organisms at low concentrations to alter the pathogenicity of the entire community, targeted therapy towards such constituents may generate clinically relevant ecological disturbances.
The advantage of characterizing microbial communities using culture-independent molecular methodologies (based on the universally present 16S rRNA gene) include the ability to assess for those organisms that have been killed as a result of prior antibacterial therapy or during transportation/sampling and to identify those organisms that are fastidious or are uncultivable using routine microbiologic practices. However, inherent to such approaches is the fact that they identify nucleic acid belonging to an organism and cannot predict viability at the time of sampling. Therefore, the biological interactions that contribute to community pathogenesis are not always clear. Novel approaches are increasingly being used to more effectively discern the composition of live organisms during microbial profiling [65, 66]; application of these towards the future characterization of the pyogenic infections will undoubtedly generate valuable data. Additionally, without the ability to grow and pass bacteria in the presence of antibacterial agents, vital antibiogram data are not available for clinical use.
There are a number of limitations of our study. This study was meant as an initial foray into the area of polymicrobial infections and, as such, we chose to sample three distinct pyogenic infections, each with a relatively small number of samples. This small sample size increases the risk of bias. The clinical significance of the polymicrobial nature of infections at these sites requires further investigation. Low-level bacterial DNA contamination has been demonstrated to exist in a number of the reagents for pyrosequencing [67, 68]. As such, sterile samples, when subjected to deep sequencing, have often identified low levels of bacterial DNA [56]. However, in clinical situations whereby high bacterial concentrations are expected, such as pus, low-level contamination would not be a significant issue and is minimized by using stringent criteria such as the analysis of only specimens with at least 1,000 sequence reads and the inclusion of only those organisms accounting for >1 % of the total sequence reads. Our assumptions of intrinsic resistance based on genus identification, while relevant for most members of a genus/family, have several notable exceptions (for example, Proteus mirabilis, unlike other members of the tribe Proteeae, does not produce an inducible AmpC β-lactamase). Furthermore, the relatively small number of organisms intrinsically resistant to therapies and the relatively small sample size for each cohort makes small-effect observations impossible, particularly with cross-sectional data. Most importantly, acquired resistance mechanisms, which account for the bulk of bacterial resistant determinants [69–71], are not assessed by the approaches that we have utilized.
From this data, we can suggest that molecular-based technologies yield a much more complex perspective on the microbial etiology of pyogenic infections. Clearly, traditional aerobic and anaerobic microbial culture-based approaches underestimate the true diversity of organisms in these infections. However, our data suggests that, in the majority of cases, the numerically dominant organism is cultivated and correctly identified by routine microbiologic practices. The additional information provided by the application of advanced culture-independent technologies affords an invaluable perspective on the invasive nature of microbial communities. Characterizing the dynamic changes in microbial constituents, microbe–microbe interactions, and the interplay of the immune system within these communities over time represents a promising area for future research.
References
Laupland KB, Ross T, Church DL et al (2006) Population-based surveillance of invasive pyogenic streptococcal infection in a large Canadian region. Clin Microbiol Infect 12:224–230
Xiao F, Tseng MY, Teng LJ et al (2005) Brain abscess: clinical experience and analysis of prognostic factors. Surg Neurol 63:442–449, discussion 449–50
Roche M, Humphreys H, Smyth E et al (2003) A twelve-year review of central nervous system bacterial abscesses; presentation and aetiology. Clin Microbiol Infect 9:803–809
Chen SC, Tsai SJ, Chen CH et al (2008) Predictors of mortality in patients with pyogenic liver abscess. Neth J Med 66:196–203
Kaplan GG, Gregson DB, Laupland KB (2004) Population-based study of the epidemiology of and the risk factors for pyogenic liver abscess. Clin Gastroenterol Hepatol 2:1032–1038
Alvarez Pérez JA, González JJ, Baldonedo RF et al (2001) Clinical course, treatment, and multivariate analysis of risk factors for pyogenic liver abscess. Am J Surg 181:177–186
Rahman NM, Chapman SJ, Davies RJ (2006) The approach to the patient with a parapneumonic effusion. Clin Chest Med 27:253–266
Wolcott RD, Gontcharova V, Sun Y et al (2009) Bacterial diversity in surgical site infections: not just aerobic cocci any more. J Wound Care 18:317–323
Dowd SE, Sun Y, Secor PR et al (2008) Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43
Dowd SE, Wolcott RD, Sun Y et al (2008) Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS One 3:e3326
Al Masalma M, Armougom F, Scheld WM et al (2009) The expansion of the microbiological spectrum of brain abscesses with use of multiple 16S ribosomal DNA sequencing. Clin Infect Dis 48:1169–1178
Al Masalma M, Lonjon M, Richet H et al (2012) Metagenomic analysis of brain abscesses identifies specific bacterial associations. Clin Infect Dis 54:202–210
Sibley CD, Rabin H, Surette MG (2006) Cystic fibrosis: a polymicrobial infectious disease. Future Microbiol 1:53–61
Sibley CD, Parkins MD, Rabin HR et al (2008) A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci U S A 105:15070–15075
Sibley CD, Grinwis ME, Field TR et al (2010) McKay agar enables routine quantification of the ‘Streptococcus milleri’ group in cystic fibrosis patients. J Med Microbiol 59:534–540
Sibley CD, Grinwis ME, Field TR et al (2011) Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One 6:e22702
Clinical and Laboratory Standards Institute (CLSI) (2007)Methods for antimicrobial susceptibility testing of anaerobic bacteria; Approved Standard—Seventh edition. CLSI, Wayne, PA
Clinical and Laboratory Standards Institute (CLSI) (2008) Interpretive criteria for identification of bacteria and fungi by DNA target sequencing; Approved Guideline. CLSI, Wayne, PA
Clinical and Laboratory Standards Institute (CLSI) (2011)Performance standards for antimicrobial susceptibility testing; Twenty-first informational supplement. CLSI, Wayne, PA
Dowd SE, Sun Y, Wolcott RD et al (2008) Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis 5:459–472
Dowd SE, Callaway TR, Wolcott RD et al (2008) Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8:125
Gontcharova V, Youn E, Wolcott RD et al (2010) Black box chimera check (B2C2): a Windows-based software for batch depletion of chimeras from bacterial 16S rRNA gene datasets. Open Microbiol J 4:47–52
Dowd SE, Zaragoza J, Rodriguez JR et al (2005) Windows .NET Network Distributed Basic Local Alignment Search Toolkit (W.ND-BLAST). BMC Bioinformatics 6:93
Cole JR, Wang Q, Cardenas E et al (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145
Mandell GL (2009) Mandell, Douglas, and Bennett’s principles and practices of infectious diseases, 7th edn. Elsevier, Philadelphia
Light RW (1995) A new classification of parapneumonic effusions and empyema. Chest 108:299–301
Laupland KB, Gregson DB, Zygun DA et al (2004) Severe bloodstream infections: a population-based assessment. Crit Care Med 32:992–997
Fajardo A, Martínez-Martín N, Mercadillo M et al (2008) The neglected intrinsic resistome of bacterial pathogens. PLoS One 3:e1619
European Committee on Antimicrobial Susceptibility Testing (EUCAST) (2008) EUCAST Expert rules in antimicrobial susceptibility testing, version 1, April 2008. Available online at: http://www.srga.org/eucastwt/EUCAST%20Expert%20rules%20final%20April_20080407.pdf. Accessed 15 Nov 2011
Laupland KB, Parkins MD, Ross T et al (2007) Population-based laboratory surveillance for tribe Proteeae isolates in a large Canadian health region. Clin Microbiol Infect 13:683–688
DiBaise JK, Zhang H, Crowell MD et al (2008) Gut microbiota and its possible relationship with obesity. Mayo Clin Proc 83:460–469
Hattori M (2010) Genetic analysis of intestinal microbiome by metagenomics. Nihon Rinsho 68(Suppl 8):506–510
Wolcott RD, Gontcharova V, Sun Y et al (2009) Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol 9:226
Gontcharova V, Youn E, Sun Y et al (2010) A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. Open Microbiol J 4:8–19
Smith DM, Snow DE, Rees E et al (2010) Evaluation of the bacterial diversity of pressure ulcers using bTEFAP pyrosequencing. BMC Med Genomics 3:41
Erb-Downward JR, Thompson DL, Han MK et al (2011) Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6:e16384
Duan K, Dammel C, Stein J et al (2003) Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol Microbiol 50:1477–1491
Sibley CD, Duan K, Fischer C et al (2008) Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog 4:e1000184
Shinzato T, Saito A (1994) A mechanism of pathogenicity of “Streptococcus milleri group” in pulmonary infection: synergy with an anaerobe. J Med Microbiol 40:118–123
Wade BH, Kasper DL, Mandell GL (1983) Interactions of Bacteroides fragilis and phagocytes: studies with whole organisms, purified capsular polysaccharide and clindamycin-treated bacteria. J Antimicrob Chemother 12(Suppl C):51–62
Rotstein OD, Pruett TL, Simmons RL (1985) Mechanisms of microbial synergy in polymicrobial surgical infections. Rev Infect Dis 7:151–170
Edwards R (1997) Resistance to beta-lactam antibiotics in Bacteroides spp. J Med Microbiol 46:979–986
Brook I (2004) Beta-lactamase-producing bacteria in mixed infections. Clin Microbiol Infect 10:777–784
Brook I (2002) Microbiology of polymicrobial abscesses and implications for therapy. J Antimicrob Chemother 50:805–810
Prasad KN, Mishra AM, Gupta D et al (2006) Analysis of microbial etiology and mortality in patients with brain abscess. J Infect 53:221–227
Jansson AK, Enblad P, Sjölin J (2004) Efficacy and safety of cefotaxime in combination with metronidazole for empirical treatment of brain abscess in clinical practice: a retrospective study of 66 consecutive cases. Eur J Clin Microbiol Infect Dis 23:7–14
Carpenter J, Stapleton S, Holliman R (2007) Retrospective analysis of 49 cases of brain abscess and review of the literature. Eur J Clin Microbiol Infect Dis 26:1–11
Moore-Gillon JC, Eykyn SJ, Phillips I (1981) Microbiology of pyogenic liver abscess. Br Med J (Clin Res Ed) 283:819–821
Corredoira J, Casariego E, Moreno C et al (1998) Prospective study of Streptococcus milleri hepatic abscess. Eur J Clin Microbiol Infect Dis 17:556–560
Maskell NA, Davies CW, Nunn AJ et al (2005) U.K. controlled trial of intrapleural streptokinase for pleural infection. N Engl J Med 352:865–874
Ahmed RA, Marrie TJ, Huang JQ (2006) Thoracic empyema in patients with community-acquired pneumonia. Am J Med 119:877–883
Rahimian J, Wilson T, Oram V et al (2004) Pyogenic liver abscess: recent trends in etiology and mortality. Clin Infect Dis 39:1654–1659
Chan KS, Chen CM, Cheng KC et al (2005) Pyogenic liver abscess: a retrospective analysis of 107 patients during a 3-year period. Jpn J Infect Dis 58:366–368
Sabbaj J (1984) Anaerobes in liver abscess. Rev Infect Dis 6(Suppl 1):S152–S156
Zaura E, Keijser BJ, Huse SM et al (2009) Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol 9:259
Charlson ES, Bittinger K, Haas AR et al (2011) Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med 184:957–963
Charlson ES, Chen J, Custers-Allen R et al (2010) Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5:e15216
Ruiz-Hernández JJ, León-Mazorra M, Conde-Martel A et al (2007) Pyogenic liver abscesses: mortality-related factors. Eur J Gastroenterol Hepatol 19:853–858
Zibari GB, Maguire S, Aultman DF et al (2000) Pyogenic liver abscess. Surg Infect (Larchmt) 1:15–21
Lipsky BA (2007) Empirical therapy for diabetic foot infections: are there clinical clues to guide antibiotic selection? Clin Microbiol Infect 13:351–353
Lipsky BA, Armstrong DG, Citron DM et al (2005) Ertapenem versus piperacillin/tazobactam for diabetic foot infections (SIDESTEP): prospective, randomised, controlled, double-blinded, multicentre trial. Lancet 366:1695–1703
Solomkin J, Teppler H, Graham DR et al (2004) Treatment of polymicrobial infections: post hoc analysis of three trials comparing ertapenem and piperacillin–tazobactam. J Antimicrob Chemother 53(Suppl 2):ii51–ii57
Wacha H, Hau T, Dittmer R et al (1999) Risk factors associated with intraabdominal infections: a prospective multicenter study. Peritonitis Study Group. Langenbecks Arch Surg 384:24–32
Dupont H, Friggeri A, Touzeau J et al (2011) Enterococci increase the morbidity and mortality associated with severe intra-abdominal infections in elderly patients hospitalized in the intensive care unit. J Antimicrob Chemother 66:2379–2385
Nocker A, Richter-Heitmann T, Montijn R et al (2010) Discrimination between live and dead cellsin bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol 13:59–65
Castillo M, Martín-Orúe SM, Manzanilla EG et al (2006) Quantification of total bacteria, enterobacteria and lactobacilli populations in pig digesta by real-time PCR. Vet Microbiol 114:165–170
Grahn N, Olofsson M, Ellnebo-Svedlund K et al (2003) Identification of mixed bacterial DNA contamination in broad-range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiol Lett 219:87–91
Spangler R, Goddard NL, Thaler DS (2009) Optimizing Taq polymerase concentration for improved signal-to-noise in the broad range detection of low abundance bacteria. PLoS One 4:e7010
McGowan JE Jr (2006) Resistance in nonfermenting gram-negative bacteria: multidrug resistance to the maximum. Am J Infect Control 34:S29–S37, discussion S64–73
Boucher HW, Talbot GH, Bradley JS et al (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12
Rice LB (2006) Antimicrobial resistance in gram-positive bacteria. Am J Infect Control 34:S11–S19, discussion S64–73
Funding
This project was performed with starter grants from both Calgary Laboratory Services and the Department of Medicine Research Development Fund from the Calgary Health Zone of Alberta Health Services.
Disclosures
C.D.S., D.L.C., S.E.D., M.G.S., and M.D.P. have no conflicts to report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sibley, C.D., Church, D.L., Surette, M.G. et al. Pyrosequencing reveals the complex polymicrobial nature of invasive pyogenic infections: microbial constituents of empyema, liver abscess, and intracerebral abscess. Eur J Clin Microbiol Infect Dis 31, 2679–2691 (2012). https://doi.org/10.1007/s10096-012-1614-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-012-1614-x