
Abstract
Multiple reaction monitoring (MRM)-based targeted metabolomics can simultaneously analyze up to hundreds of metabolites with high-throughput, good reproducibility, and wide dynamic range. However, when hundreds or thousands of MRM transitions are measured with tens to hundreds of biological samples, the complexity of MRM dataset acquired is no longer amenable to manual evaluation, and presents a challenge for targeted metabolomics. Here, we developed an R package, namely MRMAnalyzer, to process large set of MRM-based targeted metabolomics data automatically without any manual intervention. To demonstrate our MRMAnalyzer program, we first developed a targeted metabolomic method that simultaneously analyzes 182 metabolites in one 15-min LC run, and demonstrated the data processing procedures using MRMAnalyzer. The data processing steps include “pseudo” accurate m/z transformation, peak detection and alignment, metabolite identification, quality control check and statistical analysis. Finally, a targeted metabolomic assay was designed and integrated with MRMAnalyzer to profile the metabolic changes in Escherichia coli subjected to the protein expression. The generated MRM dataset consisting of more than 8000 MRM transitions were readily processed using MRMAnalyzer within 20 min without any manual intervention. Fourty seven out of 140 detected metabolites, enriched in six metabolic pathways, were found significantly affected in E. coli metabolome. In summary, a targeted metabolomic platform is developed for high-throughput metabolite profiling and automated data processing, and the MRMAnalyzer program is a high efficient informatics tool for large scale targeted metabolomics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metabolome is a collection of biochemically active molecules, namely metabolites, with a high level of chemical diversity in biological systems (Nicholson and Lindon 2008; Patti et al. 2012; Rabinowitz and Silhavy 2013; Fiehn 2002). Two mass spectrometry (MS) based approaches are widely used for metabolite profiling: untargeted and targeted metabolomics, each with its advantages and disadvantages (Dettmer et al. 2007; Lenz and Wilson 2007). Untargeted (or called non-targeted) metabolomics aims to comprehensively study the metabolome at the systems level and, therefore, tries to measure all known and unknown metabolites in a given biological sample. Performing untargeted metabolomic experiments can make discoveries that bridge the gaps between gene functions, metabolic activities and biological/physiological phenotype (Fiehn 2002; Pernet et al. 2015; Chen et al. 2012). The major challenges of this approach rest with the sophisticated informatics for data processing and time and efforts required to identify and elucidate chemical structures of both known and unknown metabolites (Smith et al. 2005, 2006; Tautenhahn et al. 2011; Horai et al. 2010; Zhu et al. 2013). Many informatics tools like XCMS (Smith et al. 2006; Tautenhahn et al. 2012; Katajamaa et al. 2006), MZmine (Katajamaa et al. 2006), MetAlign (Lommen 2009) and many others have been developed for untargeted metabolomics data processing.
Targeted metabolomics has the less ambitious goal by selectively measuring a set of known essential metabolites from biologically relevant metabolic pathways (Patti et al. 2012; Dudley et al. 2010). Triple quadrupole mass spectrometry (QqQ-MS) based multiple reaction monitoring (MRM) technique is widely used for targeted metabolomics, because of its high selectivity and sensitivity, good reproducibility and wide dynamic range in quantitative analysis (Buescher et al. 2010; Kvitvang et al. 2011). MRM technique has two stages of mass selection: the selection of precursor ion in Q1 and its product ion in Q3. The appropriate selection of the precursor ion-product ion pair, referred as one MRM transition, representing a given metabolite, enables the structure specific measurement of the chosen metabolite. With the rapid development of QqQ-MS, for example, faster scan speed, higher ionization efficiency and ion transmission efficiency, it is now possible to simultaneously analyze hundreds of pre-selected metabolites with no structural/identity ambiguity. The ability of MRM based targeted metabolomics to generate highly reproducible and quantitatively accurate data enables hypothesis-driven metabolomics research. It allows consistent analyses of a set of pre-selected metabolites in different sample groups, and determines whether metabolic pathways are affected by, for example, environmental stimuli, treatment of drugs, or genetic alterations (Mazzarino et al. 2008; Locasale et al. 2012).
In recent years, many efforts were made to continuously increase the number of metabolites that can be detected with the MRM based technique. For example, in 2006, Bajad et al. reported a HILIC-MS based MRM method that can selectively measure up to 142 metabolites in total in two positive and negative analyses (Bajad et al. 2006). The run time for two analyses is around 90 min. The pre-selected 142 metabolites cover about 25 % of the known metabolome of bacteria Escherichia coli (E. coli). Later in 2010, Wei et al. developed a column switch method that employed three different columns to separate and simultaneously detect 205 targeted metabolites in a 10-min run, called multiplexed LC–MS–MRM method (Wei et al. 2010). Recently, a positive/negative polarity switching targeted metabolomics method was demonstrated by Asara et al., that 258 metabolites were measured in a single 15-min run. In all of these studies, one target metabolite has only one characteristic MRM transition monitored to increase the throughput in a single analysis (Yuan et al. 2012).
However, with >200 transitions that can be measured in a single run, the complexity of MRM data acquired in one experiment is no longer amenable to manual evaluation. For example, a typical targeted metabolomic study consists of tens to hundreds biological samples, and 100–500 transitions in each sample analysis. In addition to biological samples, various types of quality control samples such as internal standard test mixture and pooled biological samples that used to assess the overall performance of instrument or data normalization are also necessary. Manual verification of MRM dataset in a large scale is often laborious, erroneous and irreproducible (Reiter et al. 2011; Tsugawa et al. 2013). Most manual data analysis is based on ad hoc criteria, precluding the consistent data analysis across multiple samples or different biological studies, and lack of an objective and reproducible assessment of the error rates of analyses. Therefore, efforts have been made to automate MRM data processing in both proteomics and metabolomics fields. Many programs for targeted proteomics, such as Skyline (MacLean et al. 2010) and mProphet (Reiter et al. 2011), have been well developed and proven outperforming manual analyses. However, very limited programs are readily available for targeted metabolomics data analysis. Currently, MRM based targeted metabolomics data is mainly processed using the quantitative analysis software provided by instrument vendors, such as MultiQuant from AB Sciex, MassHunter from Agilent. Each MS vendor has its own software, and limits to process the data acquired from instruments sold by the specific vendor. The vendor software is primarily designed for absolute quantitative analyses of chemical compounds, for example, drugs and pesticides. Processing targeted metabolomics data heavily relies on the manual verification of boundaries of peak integration, signal interference, and metabolite identification by well-trained analytical chemists. In addition, the peak alignment across multiple samples is essential for metabolomics data analysis, but not feasible with current vendor software.
The development of data processing software for high-throughput MRM based targeted metabolomics largely lags behind. Only a very few programs, such as MRMPROBS (Tsugawa et al. 2013, 2014), MMSAT (Wong et al. 2012) and MAVEN (Clasquin et al. 2012) have been developed to process MRM data for targeted metabolomics. For example, MRMPROBS requires at least two transitions for one metabolite and retention time to construct probabilistic scores for MRM transition grouping and metabolite identification. In most high-throughput targeted metabolomics study, only one MRM transition per metabolite is monitored in order to increase the throughput as much as possible in a single analysis, which makes data processing not compatible with MRMPROBS. MMSAT is able to detect MRM peak but lacks metabolite identification capability. The metabolite identification is still required by manual verification after data processing, which is laborious and often erroneous. Additionally, these programs lack the capability to fully automatically process large scale MRM dataset with thousands of MRM transitions and various sample types.
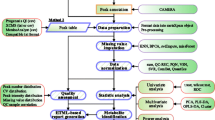
In this work, we developed an integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing. The platform combines a well-designed targeted metabolomics assay and an automated data processing R package, named as MRMAnalyzer. We first developed a MRM based targeted metabolomics method by combing hydrophilic interaction liquid chromatography (HILIC) separation and positive/negative polarity switching towards the detection of 182 polar metabolites in a single 15-min run. Further, we illustrated the newly developed MRMAnalyzer program for the automated MRM data processing including five steps such as “pseudo” accurate m/z transformation, peak detection and alignment, metabolite identification, quality control check and statistical analysis (Fig. 1). Finally, a targeted metabolomic assay was developed and integrated with the MRMAnalyzer for automated data analysis to study the metabolomic changes in E. coli under stress of the protein expression.

The integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing using the MRMAnalyzer program
2 Materials and methods
2.1 Chemicals
Chemical standards were all purchased from Sigma Aldrich (St. Louis, MO, USA) and J&K Scientific (Beijing, China). Ammonium acetate and ammonium hydroxide were purchased from Fisher Scientific (Morris Plains, NJ, USA). LC–MS grade water (H2O), methanol (MeOH) and acetonitrile (ACN) were purchased from Honeywell (Muskegon, MI, USA).
2.2 Metabolite extraction
The E. coli cell samples (OD600 nm = 1.0, 10 mL) and human cell pellets (~106 HeLa cells) were extracted using a MeOH:ACN:H2O (2:2:1, v/v) solvent mixture. A volume of 1 mL of cold solvent was added to each cell pellet, vortexed for 30 s and incubated in liquid nitrogen for 1 min. The samples were then allowed to thaw at room temperature and sonicated for 10 min. This freeze–thaw cycle was repeated three times in total. To precipitate proteins, the samples were incubated for 1 h at −20 °C, followed by 15 min centrifugation at 13,000 rpm and 4 °C. The resulting supernatant was removed and evaporated to dryness in a vacuum concentrator. The dry extracts were then reconstituted in 100 µL of ACN:H2O (1:1, v/v), sonicated for 10 min, and centrifuged 15 min at 13,000 rpm and 4 °C to remove insoluble debris. The supernatants were transferred to HPLC vials and stored at −80 °C prior to LC/MS analysis.
D. melanogaster (w 5905 strain) lines were obtained from the laboratory of Dr. Nan Liu (IRCBC, Chinese Academy of Sciences, Shanghai, China). The whole body samples of male flies aged 30 days were prepared by collecting ten flies in an Eppendorf tube and plunging them into liquid nitrogen. Then each fly sample was homogenized with 200 μL of H2O and five ceramic beads using the homogenizer (Precellys 24, Bertin Technologies, France). Then 800 μL ACN:MeOH (1:1, v/v) was added for metabolite extraction. The samples then vortexed for 30 s and sonicated for 10 min. The rest of the procedure was the same as described for E. coli and human cell samples.
Human plasma samples (200 μL, Equitech-Bio. Inc, USA) were extracted with 800 μL ACN:MeOH (1:1, v/v). The samples then vortexed for 30 s and sonicated for 10 min. The rest of the procedure was the same as described for E. coli sample and human cells.
2.3 LC–MS/MS analysis
The LC–MS analysis were performed using an HPLC system (1260 series, Agilent Technologies) coupled to a triple quadrupole mass spectrometer (Agilent 6460 QqQ, Agilent Technologies). Phenomenex Luna aminopropyl column [particle size, 3 μm; 100 mm (length) × 2.1 mm (i.d.)] was chosen for its wide pH range (1.5–11.0) and good stability in 100 % aqueous mobile phase and capability to strongly retain highly polar metabolites. The mobile phase A = 25 mM ammonium acetate and 25 mM ammonium hydroxide in 100 % water (pH ~9.75), and B = 100 % acetonitrile, were used for both in ESI positive and negative modes. The linear gradient elutes from 85 to 75 % B (0–2 min), 75 to 0 % B (2–9 min), 0 % B (9–14 min), 0 to 85 % B (14–15 min), then stays at 85 % B for 5 min. The flow rate was 0.3 mL/min and the sample injection volume was 6 μL. ESI source conditions were set as followings: sheath gas temperature, 350 °C; dry gas temperature, 350 °C; sheath gas flow, 11 L/min; dry gas flow, 10 L/min; capillary voltage, 4000 V or −3500 V in positive or negative modes, respectively; nozzle voltage, 500 V; and nebulizer pressure, 30 psi. For targeted metabolomic analyses, 182 MRM transitions representing the 182 metabolites were simultaneously monitored, and the positive/negative polarity switching was used. The dwell time for each MRM transition is 3 ms, and the total cycle time is 1.263 s.
To construct the metabolite MRM library, each metabolite standard (100 μg/mL) was first analyzed both in ESI positive and negative modes via flow injection using the software MassHunter Optimizer (Agilent Technologies) to get the optimal MRM transition parameters, such as ionization polarity, product ion, collision energy and fragmentor voltage. Then the retention time (rtsingle) of each metabolite was determined by measuring the corresponding MRM transition individually on a Luna aminopropyl column. The metabolite MRM library consists of 182 metabolites with metabolite name, ID, MRM transition parameters and retention time (rtsingle). The related information was summarized in Table S1 in the Supplementary Information.
For targeted metabolomic analyses of the E. coli metabolic response to protein expression, bacteria E. coli (BL21 strain) were transfected with plasmid pET22b for the α-synuclein expression. The samples were then divided into 36 replicates. Half of the samples (n = 18) were treated with IPTG (isopropyl-β-d-thiogalactoside, final concentration = 0.2 mM) to induce the α-synuclein expression, and the other half (n = 18) were treated with equal LB medium as controls. For each replicate, 10 mL solution (OD600 nm = 1.0) was harvested for the metabolite extraction. Pooled samples were also prepared by mixing aliquots (5 μL) of each extracted sample. Before LC–MS/MS analysis, retention time quality control sample (RTQC) was prepared by mixing ten representative metabolites. The retention times of the chosen metabolites distribute almost equally in the 15-min LC run.
2.4 MRMAnalyzer data processing
MRMAnalyzer is developed using R scripts, and is provided in Supplementary Information. The format for MRM raw data (.d files from Agilent data acquisition software MassHunter B.06.00) were first converted to files in mzML format using the “msconvert” program from ProteoWizard (version 3.0.6526). Then the mzML files were converted into text format files using the “mscat” program incorporated in ProteoWizard (version 3.0.6526) so that the data can be loaded into R and processed by MRMAnalyzer. The generated text format files contain four columns: transition index, Q1 and Q3 specifications of MRM transitions, retention time and intensity, respectively.
The MRM data processing using the MRMAnalyzer includes five steps: “pseudo” accurate m/z transformation, peak detection and alignment, metabolite identification, quality control check and statistical analysis. First, the whole MRM dataset (in text formats) was read into R and transformed as “pseudo” high resolution data so that the continuous wavelet transform (CWT) algorithm can be used for peak detection. For this purpose, each data point in one MRM (Q1/Q3) transition was assigned by a “pseudo” accurate m/z (i, i = 1.0000, 2.0000,…, or n, where n denotes the total number of transitions), thus the minimum difference in m/z between these transitions is 1 Dalton and mass accuracy within one transition is 0 ppm.
Secondly, CWT based peak detection algorithm (centWave) was used for MRM peak detection. CentWave algorithm was first developed by Tautenhahn et al. (2008) for peak detection using high resolution LC–MS data, and later was incorporated into the popular, LC/MS data analysis software, XCMS. CWT based peak detection algorithm (centWave function) is chosen mainly for its robustness against noise interferences. However, centWave function is primarily designed to process high resolution MS data only, because it relies on the detection of regions of interest (ROI) where m/z data have deviations less than a certain ppm (for example, 20 ppm for TOF data). Therefore, low resolution MRM data must be converted to “pseudo” high resolution data so it is compatible with CWT based peak detection method. In MRMAnalyzer, centWave function was slightly modified to fit MRM data processing. Specifically, for manipulation of peaks in data processing steps, data in the “mz” column of detected peaks are converted as integer type before returning. Two main parameters in MRMAnalyzer is “peakwidth” and “snthresh”. The parameter “peakwidth” for peak detection was set as c(10, 200). The parameter “peakwidth” is the chromatographic peak width, given as the range of minimum and maximum values of peak width in seconds. Here in our experiment, the peak widths of majority chromatographic peaks are about 10–20 s. But some metabolites, for examples, choline and N-Acetylputrescine, the peak width can be as large as 200 s. Thus we use the parameter c(10,200) for peak detection. The default value of parameter “snthresh” is set as 3 to sensitive peak detection. Retention time correction and peak alignment across different samples were achieved by modifying ordered bijective interpolated warping (obiwarp) algorithm previously developed by Prince et al. (2006). A standard mixture sample (182STD_mix) that contains all of the 182 metabolites was measured together with biological samples in each experiment, and was used as the reference sample for peak alignment and metabolite identification. MRM peaks detected in each biological sample were aligned to their corresponding peaks in the 182STD_mix sample.
The identification of detected peak groups was achieved by comparing the “pseudo” accurate m/z and retention time between detected peak groups and the metabolite MRM library. The “pseudo” m/z value must be exactly matched (0 ppm error), and the retention time shift tolerance is set as 60 s. In most cases, one metabolite has either no or one matched peak group in biological samples. If one metabolite has multiple matched peak groups, the metabolite ID is assigned to the peak group with the highest intensity in reference sample (i.e.,182STD_mix). The reference sample is carefully prepared and the concentration of each metabolite is optimized (Supplementary Information Table 1) so that the most intensive peak in a defined retention time region (rtsingle ± 60 s in our experiment) is the target metabolite. Here is the protocol how the reference sample (182STD_mix) has been thoroughly optimized and prepared. First, roughly 200 metabolites (2.5 µM each) were mixed as standard mixture sample and measured. Then we manually checked (1) whether each metabolite could be detected; and (2) whether the targeted metabolite peak is the most intensive peak in a defined retention time region (rtsingle ± 60 s). The results showed that 182 standards out of 200 metabolites could be detected, and 152 metabolites (2.5 µM each) have the most intensive peaks in a defined retention time region (rtsingle ± 60 s). For the other 30 metabolites, we decreased the concentrations of 3 metabolites with tailing chromatographic peaks and intensive MS responses to 1 µM to reduce potential ion suppression effect. The concentrations of the rest 27 metabolites were increased from 2.5 to 10–45 µM. The increased concentration ensures that the targeted metabolite peak becomes the most intensive peak in the defined retention time region (rtsingle ± 60 s). Finally, we prepared the reference sample (182STD_mix) with optimized concentrations of each metabolite (Supplementary Information Table 1), and each peak of 182 metabolites was manually re-checked to meet the prerequisite that the most intensive peak in a defined retention time region (rtsingle ± 60 s) is the target metabolite.
The quality control samples were processed together with the biological samples. The allowable retention time shift of metabolites in RTQC samples is 30 s compared to the MRM library. Detected metabolites in pooled samples with coefficient of variation (CV) less than 30 % were denoted as reproducible measurements.
Finally, statistical analyses between two sample groups were performed by calculating the fold changes and p values of metabolites. Welch t test with unequal variances was used to obtain p values. Metabolites with p values <0.05, fold changes >1.5, were marked as the significantly changed metabolites between sample groups. The quantitative and qualitative information of detected metabolites was output as an excel file for pathway analysis using tools such as MetaboAnalyst.
3 Results and discussion
3.1 LC–MS/MS method for targeted detection of 182 metabolites
We have developed a targeted metabolomics method based on HILIC-MS for the quantitative detection of 182 polar metabolites with good reproducibility and sensitivity. The metabolite library including metabolite name, formula, KEGG ID, MRM transitions, retention time and other information is listed in Table 1 in the Supplementary Information. The 182 polar metabolites in our analysis are selected for their significant biological functions, covering 32 KEGG metabolic pathways (Supplementary Information Table 2). MRM transition for each metabolite was optimized in both positive and negative modes to decide ionization polarity, select the product ion monitored in Q3, and choose optimized collision energy and fragmentor voltage (Supplementary Information Table 1). The optimized parameters like collision energy and fragmentor voltage are unique to the specific instrument employed and the MS parameters should be re-optimized when transferring the method to another QqQ instrument from different vendors. Potential interference between MRM transitions was taken into consideration when selecting a product ion (Q3 ion). For example, citric acid and isocitric acid have the same formula and molecular weight and cannot be chromatographically separated from each other, so the characteristic product ions of m/z 87.0 and m/z 73.0 were chosen as the targeted product ions of citric acid and isocitric acid, respectively. However, isomeric metabolites such as fumaric acid and maleic acid, glucose-6-phosphate and fructose-6-phosphate that cannot be differentiated by either retention time or product ions, are labeled as one metabolite (i.e., “Fumaric acid/Maleic acid”) in our list. Positive/negative polarity switching technique is used to increase the MS detection throughput. In the developed method, 125 positive transitions and 57 negative transitions are simultaneously detected in one experiment. The HILIC separation gradient was optimized using a standard mixture sample that contains all of the 182 metabolites (referred as182STD_mix) and tries to separate all metabolites as much as possible in 15 min (Fig. 2a). For each metabolite, the retention time is also individually measured, and marked as rtsingle in the metabolite library for identification purpose.

a Overlapped extracted ion chromatographs (EICs) for the 182 metabolites in the 182STD_mix sample. The peak intensity was normalized to 100 % for each peak. b MRM data transformation: each MRM transition (Q1/Q3) is assigned by a “pseudo” accurate m/z value ranging from 1.0000 to 182.0000. Peak detection was operated on each “pseudo” accurate m/z channel using continuous wavelet transform based centWave algorithm
3.2 MRMAnalyzer for targeted metabolomics data processing
MRMAnalyzer package is developed under the R programming environment. As illustrated in Fig. 1, the data processing steps for MRMAnalyzer include “pseudo” accurate m/z transformation, peak detection and alignment, metabolite identification, quality control check and statistical analysis.
At first, the low mass resolution MRM data were transformed to “pseudo” high mass resolution data so that CWT based peak detection algorithm can be utilized for MRM data analysis. The MRM data acquired by QqQ-MS consist of MRM transition index, retention time and ion intensity. Aligning all the MRM transitions together forms a set of 3-dimensional data that consists of MRM transitions, retention time and ion intensity, which is very similar to high mass resolution data acquired by TOF and Orbitrap instruments. Inspired by this, here, we introduced the concept of “pseudo” accurate m/z to convert the MRM data into the format of high mass resolution data (Fig. 2b). Each MRM transition (Q1/Q3) is assigned by a “pseudo” accurate m/z value corresponding to one specific metabolite. In our work, 182 MRM transitions were assigned by the “pseudo” m/z values ranging from 1.0000 to 182.0000. The minimum difference in m/z between transitions is 1 Dalton and mass accuracy within one transition is 0 ppm. The integer “pseudo” accurate m/z values used in this manuscript can be exchanged by other numerical values such as 0.1000, 0.2000,…,n. But it should be noted that “pseudo” accurate m/z values representing different MRM transitions must be different values. The new 3-dimensional dataset that consists of “pseudo” accurate m/z, retention time and ion intensity could represent the original MRM data, and is also compatible with CWT based peak detection method. More important, the data transformation principle demonstrated here ensures that our MRMAnalyzer program can process MRM data acquired from any QqQ-MS instrument or under any experimental conditions.
After MRM data transformation, peak detection was operated on each “pseudo” accurate m/z channel using a recently introduced CWT based peak detection algorithm (centWave, Fig. 2b). centWave algorithm was first developed by Tautenhahn et al. (2008) and requires high resolution LC–MS data. We chose centWave algorithm for peak detection mainly for its robustness against noise interferences. The results show that multiple peaks are readily detected in each transition for the real biological samples. Taken one E. coli sample as an example, as many as 2046 peaks were detected in 182 MRM transitions, ranging from 0 to 39 peaks per MRM transition (Supplementary Information Fig. 1). Among these transitions, 29 of 182 transitions have no peak detection. However, the majority of transitions have more than one peak detected, highlighting the sensitivity of peak detection algorithm.
Later, peak alignment and grouping were performed across multiple biological samples. A mixture of 182 metabolite standards (referred as 182STD_mix) was measured together with biological samples, and used as the reference sample for retention time correction and peak alignment (Fig. 3a). MRM peaks detected in each biological sample were aligned to their corresponding peaks in 182STD_mix sample (Fig. 3b). Therefore, each peak group consists one peak from 182STD_mix sample and multiple MRM peaks detected from biological samples. The detected 182STD_mix peak is used for identification purpose while the MRM peaks in biological samples are used for quantitative and statistical analyses. After peak alignment and peak grouping, most interference peaks in biological samples are removed. Only 795 MRM peak groups were detected in E. coli bacteria samples, ranging from 0 to 22 peak groups per transition (Supplementary Information Fig. 2). Among the 182 MRM transitions, 17 transitions have no peak group detection, 38 transitions have one peak group detected, and the rest 127 transitions have more than one peak group detected.

a Retention time correction based on the reference sample (182STD_mix). A mixture of 182 metabolite standards (182STD_mix) was measured together with E. Coli samples and used as the reference sample for retention time correction (dash line). b MRM peak alignment and grouping using the reference sample (182STD_mix). A set of MRM peaks detected in E. coli samples (pseudo accurate m/z: 153.000) are aligned to the corresponding MRM peak observed in the reference sample (182STD_mix). Each peak group consists one peak from 182STD_mix sample and multiple MRM peaks detected from biological samples (n = 18)
Finally, the metabolite identification step is followed by matching the metabolite in the metabolite library to the detected 795 MRM peak groups. The metabolite library consists of metabolite name, MRM transitions and corresponding “pseudo” accurate m/z values, and retention time (Supplementary Information Table 1). The “pseudo” accurate m/z value must be exactly matched (0 ppm error), while the retention time shift tolerance is set as rtsingle ± 60 s (Fig. 4a). For the 18 replicates of bacteria samples mentioned above, 45 out of 182 metabolites has no matched peak groups, therefore these metabolites are not detected in these biological samples. The other 137 metabolites have at least one matched groups. Then the metabolite is assigned to the peak group with highest MRM peak intensity detected in the reference sample (i.e., 182STD_mix). As a result, 137 metabolites were identified in the 18 replicates of E. coli samples (Fig. 4b). Peak groups having no matched metabolites are signal interferences in the biological samples. This metabolite identification procedure requires a prerequisite that the most intensive MRM peak in a defined retention time region (rtsingle ± 60 s) in the 182STD_mix reference sample is the target metabolite. During the preparation of 182STD_mix sample, the concentration of each metabolite is comprehensively optimized and manually inspected to satisfy this prerequisite (see Sect. 2). The accurate assignment of metabolite ID to the peaks detected in reference sample was achieved by comparing the detected peaks in reference sample with the MRM peaks measured in single metabolite standard, and at least two qualifier transitions per single standard have been monitored to get the reliable assignment. As shown in Fig. 4a, there are interference peaks detected in biological samples (RT = 725 s, black lines). However, no corresponding peak detected in reference sample (182STD_mix). Next, the developed targeted metabolomics method and the MRMAnalyzer data processing workflow was applied on the targeted analyses of four different types of biological samples, including bacteria (E. coli), fruit fly (D. melanogaster), human cell lines (HeLa cells) and human plasma, and 115–139 metabolites were detected in different biological samples (Fig. 4b). The results demonstrated that the technique can be used to profile a variety of biological samples with different metabolite coverage.

a The metabolite identification using MRMAnalyzer: each of the 182 metabolites in MRM library was matched to the detected peak groups by comparing their “pseudo” accurate m/z value and retention time. The “pseudo” accurate m/z value tolerance is set as 0 ppm, and the retention time shift tolerance is set as 60 s. b The number of metabolites detected in D. melanogaster, bacteria E. coli, HeLa cells, and human plasma samples using the developed targeted metabolomic method combined with the MRMAnalyzer data processing
In this work, the incorporation of 182STD_mix used as the reference sample for peak alignment and metabolite identification can largely reduce the laborious work and errors on metabolite identification compared with manual verification. For example, the “Find by MRM” function in MassHunter software (version B.06.00) is the most popular way to extract and identify MRM peaks across multiple samples in targeted metabolomics. But there are issues with MassHunter software (or other vendor software) that preclude the consistent and automated data analysis of large scale datasets across multiple samples or different biological studies. For example, in MRM-based targeted metabolomics, more than one peak are often observed in one MRM transition across the chromatograph (Supplementary Information Fig. 3a). As illustrated in Supplementary Information Fig. 3a, using the “Find by MRM” function in MassHunter, the targeted metabolite ID was by default assigned to the most abundant peak in the MRM transition across the chromatograph. As a result, metabolite GDP (guanosine diphosphate, Q1/Q3 = 444.1/152.1) was assigned to the most abundant peak (RT = 740 s) in one sample, while assigned to another abundant peak (RT = 610 s) in another sample, generating inconsistent data analyses. In reality, the examples given in Supplementary Information Fig. 3 happen quite often due to unexpected interferences and complexity of biological samples. To solve this issue, one can manually verify retention times of each detected peak to correct the wrong assignments, but it is too laborious for a large scale dataset. Our MRMAnalyzer program readily solves this issue through peak alignment. All MRM peaks detected in each biological sample were aligned to their corresponding peaks in reference sample (182STD_mix). As shown in Fig. 3b (Supplementary Information), the MRM peaks at 740 s in different samples were successfully aligned to the standard MRM peak in reference sample (182STD_mix), and subsequently identified as GDP. The interference peak at 610 s has no corresponding peak in reference sample, therefore discarded as interferences by the program.
Sometimes manual verification based on retention time is not reliable due to non-linear and irreproducible shift of retention time across different samples. As illustrated in Fig. 4 (Supplementary Information), two MRM peaks were detected in one transition channel (Q1/Q3 = 176.2/70.2) within retention time rtsingle ± 60 s. The targeted metabolite citrulline has a retention time of 376 s in RT library. During manual analysis, the interference peak group 1 with higher intensity and smaller retention time shift (4 s compared to RT library) is usually mistakenly assigned as citrulline. With the incorporation of reference sample (182STD_mix) for peak alignment and identification, the interference peaks (peak group 1) in biological samples are removed after peak alignment, and the less abundant peaks (peak group 2) were correctly identified as citrulline. Each metabolite peak in reference sample was carefully manually verified during method development and optimization to ensure 100 % accuracy of identification. Therefore, our program can readily process large scale of MRM dataset in an effective way and ensure that the detected peaks are identified accurately. However, the low abundant peaks are still suggested to be further verified manually for accurate peak integration, and a more interactive program interface will be more helpful and will allow users to manually verify the peak integration in the future.
3.3 Targeted metabolomic assay for metabolic profiling
We developed a targeted metabolomic assay that combines high-throughput metabolite profiling and automated data processing using MRMAnalyzer, and demonstrated its utility for metabolite profiling of large sets of biological samples. Several quality control features are incorporated with the assay and compatible with MRMAnalyzer program for data analysis. We performed the targeted metabolomic analyses of E. coli subjected to the expression of α-synuclein protein. α-synuclein, an abundant and conserved presynaptic brain protein, is implicated as a critical factor in Parkinson’s disease (PD) (Mezey et al. 1998). Two groups of bacteria samples, with and without the expression of α-synuclein, were analyzed and each group has 18 biological replicates as a demonstration.
The targeted metabolomic assay was designed as shown in Fig. 5. Pooled samples were prepared by mixing aliquots of all the biological samples, and used as quality control (QC) samples to evaluate the stability and reproducibility of the instrument. At the beginning of a batch, three pooled samples are used for column conditioning, and the pooled samples are injected at a regular interval (every nine samples) throughout the assay analysis. After pooled samples, a retention time quality control (RTQC) sample is introduced to ensure that the retention time shift is within 30 s compared to the metabolite library. The RTQC sample contains ten metabolites, and the retention time of each metabolite distributes almost equally in the 15-min LC run (Supplementary Information Fig. 5). If retention time shifts more than 30 s, the metabolite identification may fail during data processing. In our bacteria analyses (roughly 22 h in total), the retention time shift is within 10 s compared to the MRM library (Supplementary Information Fig. 6). The RTQC samples were analyzed three times at the beginning, in the middle, and at the end of a batch, respectively. A 182STD_mix reference sample is analyzed once in the middle of the batch, and used as reference for peak alignment and metabolite identification. In addition, all biological samples were randomized and placed into the run sequence that helps to eliminate the viability in analysis caused by the run order. The MRMAnalyzer program is capable to automatically and simultaneously process RTQC samples, pooled samples and 182STD_mix reference sample and ensures passing requirements on quality control before processing the biological sample data. The processing of the whole MRM dataset that consists of 36 biological samples, five pooled samples, three RTQCs, and one 182STD_mix (more than 8000 MRM transitions in total) using the MRMAnalyzer program takes less than 20 min.

Targeted metabolomic analysis of E. coli metabolome changes in response to protein α-synuclein expression. a Run order sequence for the designed targeted metabolomic assay. b 140 metabolites were detected in E. coli samples. 47 metabolites were significantly changed with the expression of α-synuclein (p value < 0.05, and fold change >1.5). Six metabolic pathways are significantly affected by the expression of α-synuclein
All acquired MRM data were converted and analyzed by MRMAnalyzer program, and results were summarized in Fig. 5b. 140 metabolites were detected in the E. coli metabolome with our targeted metabolomic method. For each of detected metabolite, the quantitative and qualitative information was output as an excel file (Supplementary Information Table 3). The quantitative information includes metabolite intensity in each sample, fold change, and p value between two groups, while the qualitative information includes metabolite name, KEGG ID, and HMDB ID. 47 of 140 metabolites were determined as significantly changed with p value less than 0.05 and fold change higher than 1.5. The other detected 93 metabolites remain unaffected by the protein expression. The significantly changed 47 metabolites were further submitted to pathway enrichment analysis using MetaboAnalyst (Xia et al. 2009; Xia and Wishart 2011). The enrichment analysis demonstrated that six pathways including pyrimidine metabolism, protein biosynthesis, and citric acid cycle are significantly affected by the expression of α-synuclein (p < 0.05) in E. coli metabalome (Supplementary Information Table 4). Protein biosynthesis related that metabolites such as lysine, arginine and histidine are up-regulated with the expression of α-synuclein. Energy metabolism related metabolites in citric acid cycle pathway such as NADH and succinic acid were also up-regulated with the expression of α-synuclein.
4 Concluding remarks
In summary, we developed a targeted metabolomic platform for the quantitative detection of 182 polar metabolites in a single 15-min LC–MS/MS run, and demonstrated an R package, MRMAnalyzer for the automated data processing. The MRMAnalyzer program can quantitatively detect and identify metabolites from the low resolution MRM data through five processing steps, including “pseudo” accurate m/z transformation, peak detection and alignment, metabolite identification, quality control check and statistical analysis. The data transformation strategy by assigning each MRM transition to a “pseudo” accurate m/z ensures that the MRMAnalyzer program can process MRM data acquired from any QqQ-MS instrument or any experimental condition. Further, a reference sample that contains all the targeted 182 metabolites was measured together with biological samples and used for peak alignment and metabolite identification, that can effectively reduce the laborious work and errors on metabolite identification compared with manual verification. Finally, a well-designed targeted metabolomics assay that consists of retention time quality control samples (RTQC), quality control pooled samples, and biological samples was integrated with the MRMAnalyzer program, and applied to characterize the metabolic changes in bacteria E. coli under the stress of protein expression. In conclusion, the developed MRMAnalyzer program could automatically and efficiently process large scale MRM dataset, and is capable to find dysregulated metabolites and related metabolic pathways. We hope it is a valuable tool for researchers to facilitate the respective biological studies using targeted metabolomics.
References
Bajad, S. U., Lu, W. Y., Kimball, E. H., Yuan, J., Peterson, C., & Rabinowitz, J. D. (2006). Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. Journal of Chromatography A, 1125(1), 76–88. doi:10.1016/j.chroma.2006.05.019.
Buescher, J. M., Moco, S., Sauer, U., & Zamboni, N. (2010). Ultrahigh performance liquid chromatography-tandem mass spectrometry method for fast and robust quantification of anionic and aromatic metabolites. Analytical Chemistry, 82(11), 4403–4412. doi:10.1021/Ac100101d.
Chen, Q., Park, H. C., Goligorsky, M. S., Chander, P., Fischer, S. M., & Gross, S. S. (2012). Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PLoS ONE, 7(6), e37149. doi:10.1371/journal.pone.0037149.
Clasquin, M. F., Melamud, E., & Rabinowitz, J. D. (2012). LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Current Protocols Bioinformatics, 14(Unit14), 11. doi:10.1002/0471250953.bi1411s37.
Dettmer, K., Aronov, P. A., & Hammock, B. D. (2007). Mass spectrometry-based metabolomics. Mass Spectrometry Reviews, 26(1), 51–78. doi:10.1002/Mas.20108.
Dudley, E., Yousef, M., Wang, Y., & Griffiths, W. J. (2010). Targeted metabolomics and mass spectrometry. Advances in Protein Chemistry and Structural Biology, 80, 45–83. doi:10.1016/S1876-1623(10)80002-1.
Fiehn, O. (2002). Metabolomics—The link between genotypes and phenotypes. Plant Molecular Biology, 48(1–2), 155–171. doi:10.1023/A:1013713905833.
Horai, H., Arita, M., Kanaya, S., Nihei, Y., Ikeda, T., Suwa, K., et al. (2010). MassBank: A public repository for sharing mass spectral data for life sciences. Journal of Mass Spectrometry, 45(7), 703–714. doi:10.1002/jms.1777.
Katajamaa, M., Miettinen, J., & Oresic, M. (2006). MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics, 22(5), 634–636. doi:10.1093/bioinformatics/btk039.
Kvitvang, H. F. N., Andreassen, T., Adam, T., Villas-Boas, S. G., & Bruheim, P. (2011). Highly sensitive GC/MS/MS method for quantitation of amino and nonamino organic acids. Analytical Chemistry, 83(7), 2705–2711. doi:10.1021/Ac103245b.
Lenz, E. M., & Wilson, I. D. (2007). Analytical strategies in metabonomics. Journal of Proteome Research, 6(2), 443–458. doi:10.1021/Pr0605217.
Locasale, J. W., Melman, T., Song, S., Yang, X., Swanson, K. D., Cantley, L. C., et al. (2012). Metabolomics of human cerebrospinal fluid identifies signatures of malignant glioma. Molecular & Cellular Proteomics, 11(6), M111 014688, doi:10.1074/mcp.M111.014688.
Lommen, A. (2009). MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Analytical Chemistry, 81(8), 3079–3086. doi:10.1021/Ac900036d.
MacLean, B., Tomazela, D. M., Shulman, N., Chambers, M., Finney, G. L., Frewen, B., et al. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics, 26(7), 966–968. doi:10.1093/bioinformatics/btq054.
Mazzarino, M., de la Torre, X., & Botre, F. (2008). A screening method for the simultaneous detection of glucocorticoids, diuretics, stimulants, anti-oestrogens, beta-adrenergic drugs and anabolic steroids in human urine by LC-ESI-MS/MS. Analytical and Bioanalytical Chemistry, 392(4), 681–698. doi:10.1007/s00216-008-2292-5.
Mezey, E., Dehejia, A., Harta, G., Papp, M. I., Polymeropoulos, M. H., & Brownstein, M. J. (1998). Alpha synuclein in neurodegenerative disorders: Murderer or accomplice? Nature Medicine, 4(7), 755–757. doi:10.1038/Nm0798-755.
Nicholson, J. K., & Lindon, J. C. (2008). Systems biology—Metabonomics. Nature, 455(7216), 1054–1056. doi:10.1038/4551054a.
Patti, G. J., Yanes, O., & Siuzdak, G. (2012). Metabolomics: The apogee of the omics trilogy. Nature Reviews Molecular Cell Biology, 13(4), 263–269. doi:10.1038/Nrm3314.
Pernet, C., Munoz, J., & Bessis, D. (2015). PENS (papular epidermal nevus with “skyline” basal cell layer). Annales de Dermatologie et de Venereologie, 142(1), 41–45. doi:10.1016/j.annder.2014.09.003.
Rabinowitz, J. D., & Silhavy, T. J. (2013). Systems biology: Metabolite turns master regulator. Nature, 500(7462), 283–284. doi:10.1038/nature12544.
Reiter, L., Rinner, O., Picotti, P., Huttenhain, R., Beck, M., Brusniak, M. Y., et al. (2011). mProphet: automated data processing and statistical validation for large-scale SRM experiments. Nature Methods, 8(5), 430–435. doi:10.1038/nmeth.1584.
Smith, C. A., O’Maille, G., Want, E. J., Qin, C., Trauger, S. A., Brandon, T. R., et al. (2005). METLIN: a metabolite mass spectral database. Therapeutic Drug Monitoring, 27(6), 747–751.
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R., & Siuzdak, G. (2006). XCMS: Processing mass spectrometry data for metabolite profiling using Nonlinear peak alignment, matching, and identification. Analytical Chemistry, 78(3), 779–787. doi:10.1021/Ac051437y.
Tautenhahn, R., Bottcher, C., & Neumann, S. (2008). Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics,. doi:10.1186/1471-2105-9-504.
Tautenhahn, R., Cho, K., Uritboonthai, W., Zhu, Z. J., Patti, G. J., & Siuzdak, G. (2012). An accelerated workflow for untargeted metabolomics using the METLIN database. Nature Biotechnology, 30(9), 826–828. doi:10.1038/Nbt.2348.
Tautenhahn, R., Patti, G. J., Kalisiak, E., Miyamoto, T., Schmidt, M., Lo, F. Y., et al. (2011). metaXCMS: second-order analysis of untargeted metabolomics data. Analytical Chemistry, 83(3), 696–700. doi:10.1021/ac102980g.
Tsugawa, H., Arita, M., Kanazawa, M., Ogiwara, A., Bamba, T., & Fukusaki, E. (2013). MRMPROBS: A data assessment and metabolite identification tool for large-scale multiple reaction monitoring based widely targeted metabolomics. Analytical Chemistry, 85(10), 5191–5199. doi:10.1021/Ac400515s.
Tsugawa, H., Kanazawa, M., Ogiwara, A., & Arita, M. (2014). MRMPROBS suite for metabolomics using large-scale MRM assays. Bioinformatics, 30(16), 2379–2380. doi:10.1093/bioinformatics/btu203.
Wei, R., Li, G. D., & Seymour, A. B. (2010). High-throughput and multiplexed LC/MS/MRM method for targeted metabolomics. Analytical Chemistry, 82(13), 5527–5533. doi:10.1021/Ac100331b.
Wong, J. W., Abuhusain, H. J., McDonald, K. L., & Don, A. S. (2012). MMSAT: Automated quantification of metabolites in selected reaction monitoring experiments. Analytical Chemistry, 84(1), 470–474. doi:10.1021/ac2026578.
Xia, J. G., Psychogios, N., Young, N., & Wishart, D. S. (2009). MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Research, 37, W652–W660. doi:10.1093/Nar/Gkp356.
Xia, J. G., & Wishart, D. S. (2011). Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nature Protocols, 6(6), 743–760. doi:10.1038/nprot.2011.319.
Yuan, M., Breitkopf, S. B., Yang, X. M., & Asara, J. M. (2012). A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nature Protocols, 7(5), 872–881. doi:10.1038/nprot.2012.024.
Zhu, Z. J., Schultz, A. W., Wang, J. H., Johnson, C. H., Yannone, S. M., Patti, G. J., et al. (2013). Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nature Protocols, 8(3), 451–460. doi:10.1038/nprot.2013.004.
Acknowledgments
We thank the financial support provided by the startup funding from Interdisciplinary Research Center on Biology and Chemistry (IRCBC), Chinese Academy of Sciences (CAS). Z.-J. Z. is also supported by Thousand Youth Talents Program (The Recruitment Program of Global Youth Experts from Chinese government). The project is also financially supported by Agilent Technologies Thought Leader Award.
Compliance with ethical requirements
All institutional and national guidelines for the care and use of biological samples were followed. The data acquired were in accordance with appropriate ethical requirements.
Conflict of interest
There are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yuping Cai and Kai Weng have contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cai, Y., Weng, K., Guo, Y. et al. An integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing. Metabolomics 11, 1575–1586 (2015). https://doi.org/10.1007/s11306-015-0809-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-015-0809-4