
Abstract
The type and use of quality control (QC) samples is a ‘hot topic’ in metabolomics. QCs are not novel in analytical chemistry; however since the evolution of using QCs to control the quality of data in large scale metabolomics studies (first described in 2011), the need for detailed knowledge of how to use QCs and the effects they can have on data treatment is growing. A controlled experiment has been designed to illustrate the most advantageous uses of QCs in metabolomics experiments. For this, samples were formed from a pool of plasma whereby different metabolites were spiked into two groups in order to simulate biological biomarkers. Three different QCs were compared: QCs pooled from all samples, QCs pooled from each experimental group of samples separately and QCs provided by an external source (QC surrogate). On the experimentation of different data treatment strategies, it was revealed that QCs collected separately for groups offers the closest matrix to the samples and improves the statistical outcome, especially for biomarkers unique to one group. A novel quality assurance plus procedure has also been proposed that builds on previously published methods and has the ability to improve statistical results for QC pool. For this dataset, the best option to work with QC surrogate was to filter data based only on group presence. Finally, a novel use of recursive analysis is portrayed that allows the improvement of statistical analyses with respect to the ratio between true and false positives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Quality control (QC) is a ‘hot topic’ in metabolomics and many research articles have been published that both exemplify the reasons for and proposed uses of QC samples; their inclusion in metabolomics analyses is now routine in many applications (Gika et al. 2007, 2012; Guy et al. 2008; Kamleh et al. 2012). The analysis of QC samples in metabolomics was first described in 2006, although the concept of QC in validated methods has long been described (Sangster et al. 2006). In any case, QC samples are analysed at the start and end of an analytical run as well as at intermittent points throughout, and their function is to monitor the performance of a method. In a validated method, the QC sample usually consists of the blank matrix spiked with known concentrations of the analyte in order to confirm reliability of its quantification in samples throughout an analytical run (Sangster et al. 2006). In the case of metabolomics, this type of QC is not applicable since the approach is often untargeted, involving 100–1,000s of unknown compounds at unknown concentrations. Instead, the idea of analysing a ‘biological QC’ arose from the description of pooling aliquots of all samples and injecting this mixture as the QC. Since it was proposed in 2006 (Sangster et al. 2006), it is now a routine approach in metabolomics to inject this QC pool several times at the start of the analytical run to ensure system stability and to analyse the same pool at intervals throughout the analysis for monitoring purposes.
There are many ways to deal with the information provided by the intermittently injected QC pool and new methods to deal with data are constantly evolving. On interpreting the QC samples, one can decide whether or not the analysis was performed in a reliable and reproducible manor, and whether or not all/some of the data is of high enough quality to consider in statistical analyses. One common way to show this reproducibility is through the clustering of QC samples in principal components analysis (PCA). QC samples are expected to cluster closely to each other in the unsupervised plot (Sangster et al. 2006). Another measure of reproducibility is the assessment of % relative standard deviation (RSD) of each metabolic feature across QC samples (Godzien et al. 2013a).
More recently, the use of QCs has progressed to control the quality of data in large scale studies. For example, procedures for large-scale metabolomics with respect to how to utilise QC samples in data-treatment have been proposed (Dunn et al. 2011). This saw the introduction of a surrogate QC sample in metabolomics studies where the first batches of samples were analysed analytically before the collection of all samples in the cohort. In this way the QC sample still functioned as a measure of instrument stability over time and enabled final integration of data from all analytical batches; however since the sample was external to the biological samples, specific strategies for data filtering and signal correction based on QCs were proposed. In this case, it was reported that data should be filtered based on at least 50 % presence in QCs and exhibiting RSD lower than 20 % (HPLC–MS data) or 30 % (GC–MS data). Such data can then be used in batch-matching and signal correction (between filter by presence and filter by reproducibility) in order to generate a complete dataset for statistical analyses (Dunn et al. 2011). Additional uses of QCs in batch control for metabolomics studies have also been recently reported including the determination of day-to-day reproducibility in metabolite profiling (Gika et al. 2012).
Different types of QC samples have been proposed beyond the classical pooling technique and the utilisation of a surrogate QC in large-scale studies (or in studies where sample volumes do not permit the pooling of biological samples to form the QC sample) (Ciborowski et al. 2012b; Godzien et al. 2013b). For example, in a study published in 2009, three different ways to prepare QCs were exemplified that included the injection of Milli-Q water, a mixture of metabolite standards and the re-injection of some of the biological samples (Llorach et al. 2009).
Although the use of QCs is now routine in metabolomics studies and there are different strategies published for the type of sample and the way they are used in data treatment, the effect of using different QCs and the data treatment procedures that are most appropriate based on the type of QC used in an experiment is not well defined. To this end, we have designed a controlled experiment utilising HPLC–MS, whereby different metabolites have been spiked at different concentrations into human plasma in order to simulate two biological groups of samples. Moreover, other metabolites have been altered in silico to represent features that are present in one group and completely absent in the other. These types of features are usually disease or treatment specific and are either absent or present in case subjects relative to controls. QCs were formed either by pooling equal volumes of all samples into one QC (QC pool), pooling equal volumes of samples from the each group separately (QCA and QCB), or employing a surrogate QC from different human plasma (QC surrogate). The appropriateness of different filtering strategies were compared and contrasted for each with respect to their effect on the final statistical results.
The choices made in data treatment procedures have different influences on the interpretation of metabolomics data. In some studies, it is possible to choose the type of QCs to be used, however the experimental design may lead to only one possibility. In any case, different decisions can be made in data treatment and the effects of such decisions are presented herein.
2 Materials and methods
2.1 Chemicals and reagents
Ultrapure water, used to prepare all the aqueous solutions was obtained “in-house” from a Milli-Qplus185 system (Millipore, Billerica, MA, USA). LC–MS grade acetonitrile and analytical grade formic acid were purchased from Fluka Analytical (Sigma-Aldrich Chemie GmbH, Steinheim, Germany). All spiked metabolites were acquired from Sigma-Aldrich (Chemie GmbH, Germany).
2.2 Biological samples
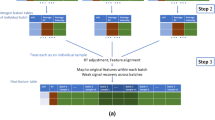
The pool of plasma was deproteinised by the use of cold methanol:ethanol 1:1 (−20 °C) in the proportion 1:3 (Ciborowski et al. 2010). Plasma was vortex-mixed for 1 min and then incubated on ice for 10 min. Vortex-mixed plasma was centrifuged at 16,000×g for 20 min at 4 °C. The resulting supernatant was collected and separated into two tubes (A and B), to which eight metabolites were added. Hydrocortisone, tetradecylamine and octanoyl-carnitine were spiked to A and octylamine, palmitoyl-carnitine and progesterone were spiked to B at different concentrations. Acetyl-carnitine and decanoyl-carnitine were spiked to both mixes in equal concentrations to control the procedure. Each metabolite was added at a concentration of 10 ppm. Samples were formed by mixing different proportions of plasma A and plasma B to simulate two groups with intra-group biological variation as described in Table 1. QCs were formed by pooling equal volumes of each sample (QC pool); equal volumes of samples from group A (QC A); equal volumes of samples from group B (QC B) or by pooling surrogate plasma that was extracted in the same way. Figure 1 shows a schematic of how samples and QCs were formed.

Sample formation. Plasma was pooled, extracted and separated into two tubes (A and B). Unique metabolites were spiked to each plasma pool (in addition to metabolites spiked to each in equal concentrations). Different proportions of plasmas A and B were mixed to form samples and QCs were formed by pooling specific samples as shown. QC surrogate was formed from a different pool of plasma that was extracted in the same way
2.3 Analysis by HPLC–QTOF–MS
The analysis was set up in one batch of samples that started with ten QC pool samples for equilibration and stabilisation of the system, followed by samples in a randomised order. After the injection of every fourth sample, one of each of the QCs were analysed until the end of the run. Eight samples were analysed for each group and five of each QC. The order of analysis is provided in the Supplementary Information (Fig. S1, SI).
All analyses were performed by HPLC–QTOF–MS (HPLC 1200 series coupled to QTOF 6520, Agilent) using a previously described method (Ciborowski et al. 2010). The system was operated with a flow rate of 0.6 mL/min and with a gradient that started with 25 % B, reached 95 % B in 35 min, returned to 25 % B in 1 min and was maintained at 25 % B for a further 9 min. Briefly, 10 μL of sample was loaded onto a reversed-phase column (Discovery HS C18 15 cm × 2.1 mm, 3 μm; Supelco) with a guard column (Discovery HS C18 2 cm × 2.1 mm, 3 μm; Supelco). The mobile phase consisted of solvents A and B: water with 0.1 % formic acid and acetonitrile with 0.1 % formic acid, respectively. Data were collected for each sample in positive ionisation modes in full scan mode (m/z 50–1,000). The capillary voltage, nebuliser gas flow rate and the scan rate were 3,000 V, 10.5 L/min and 1.02 scan/s, respectively.
2.4 Data re-processing
The molecular feature extraction (MFE) tool in Mass Hunter Qualitative Analysis (B.06.00, Agilent) was employed to clean data of background noise and to provide a list of all possible features in each sample. Features were created using the accuracy of mass measurements to group ions related to charge-state envelope, isotopic distribution, and/or the presence of adducts and dimers, as well as potential neutral loss of molecules. The MFE algorithm finds co-eluting ions that are linked and sums all ion signals into one value defined as a feature. Compound abundance is assigned as a sum of volume for each related ion/peak. Data were reprocessed considering ions such as [M+H]+ and [M+Na]+, neutral water loss and the maximum permitted charge state was double. Following this, data were aligned in Mass Profiler Professional (12.6.1, Agilent) and exported for analysis. Alignment was performed based on m/z and RT similarities within the samples. No shifts were observed in peaks over the analysis; therefore, alignment was performed without prior RT correction. Parameters applied were 1 % for RT window and 20 ppm for mass tolerance. Alignment was performed by restricting the number of ions and charge states defined previously during extraction of features.
2.5 Data pre-treatment
Data were filtered based on different criteria to compare and contrast their effects on the reproducibility of the final dataset. Data were filtered (i) based on at least 50 % presence and lower than 30 % RSD in respective QCs, where missing values were included in RSD calculation; (ii) based on at least 50 % presence and lower than 30 % RSD in respective QCs, where missing values were not included in RSD calculation and (iii) based on at least 80 % presence and lower than 30 % RSD in respective QCs, where missing values were not included in RSD calculation.
After concluding the best option for filtering data based on QC RSD, different methods of quality assurance (QA) were compared: filtered based on the selected QC RSD filtration method optimised for the present dataset from QA previously published (Dunn et al. 2011) (QAopt); QAopt with the addition of including features that were not present in QCs [quality assurance plus (QA+)] and no filtration based on QC samples. In all cases, filtering based on presence in at least n − 1 samples of at least one of the two groups was performed. For all features that passed the filters, missing values were replaced in different ways. For QC samples (n = 5) one missing value per feature was replaced by the median of all others and two missing values per feature were replaced by 1/2 of the minimum. For samples per group (n = 8) two missing values per feature were replaced by the median of all others, 3–5 missing values per feature were replaced by 1/2 of the minimum and for more than 5 missing values, they were kept as zeros.
2.6 Data treatment
Prior to statistical analysis, the distribution of data for each feature was assessed. Scripts for MATLAB (R2010a, MathWorks) were built in-house to test the normality of each feature based on the Shapiro–Wilk method and for normally distributed features, homogeneity of variance was tested based on the Levene method. For normally distributed features, p-values were calculated using the Student’s t test assuming equal variances for features that passed the Levene test and with no assumption of equal variances for all others. For non-normally distributed features, the Mann–Whitney U test was employed. Post-correction was performed for all features together using the Bonferroni method.
2.7 Recursive analysis
MFE is a naïve peak discovery tool which finds all co-eluting signals corresponding to the same molecule. Data re-processing is performed for each sample separately, therefore whenever information contained in the data file is different between samples (due to the alterations in the metabolites intensity and/or sample matrix) the final output can be slightly different. These differences cause the misassignments either for the neutral mass given or total feature intensity. Such misassignments can be sources of either false positive and/or true negative errors. Recursive analysis is based on the Find by Ion algorithm which uses previously found compounds (resulting from MFE) as the data source listed in the previous output (the list of previously found features before, after or during the data analysis procedure, saved in the format. cef—compound exchange file). Using spectra from all ions, m/z and total feature intensity are re-calculated. Data re-processing is performed for each data file separately but the search is based on the list of features created for all samples under investigation, therefore information missed in one data file can be re-covered by comparison with other samples. The workflow for recursive analysis is shown in Fig. S2, SI.
A novel application of recursive analysis was applied to features found to be statistically significantly different between groups A and B (as determined by univariate analysis). The obtained data were treated exactly the same as data obtained by MFE. To ensure conditions were comparable, Bonferroni correction was performed on equal numbers of features (Fig. S3, SI). Although recursive analysis was only performed on statistically significant features, corrected p-values were calculated simultaneously for all features (all others obtained previously by MFE). This procedure was applied to test the same number of features, since the number of masses tested affects the results of post-correction output.
3 Results and discussion
3.1 Assessment of data quality
To elucidate the effect of different QCs on the analysis and final interpretation of data, a controlled experiment has been designed. Eight metabolites were spiked into human plasma (six at varying concentrations to simulate biomarkers and two at controlled concentrations between groups) to create two biological groups, representing a biological dataset. Acquired data were checked in terms of quality and reproducibility considering pressure stability, chromatogram reproducibility, number of features per sample and total signal per sample (Godzien et al. 2013a).
Data were initially filtered based on presence in at least 90 % of one of two groups in order to assess the global analysis through PCA (Fig. 2). As can be clearly seen, the positions of different types of QCs in the PCA space were concordant with respect to the positioning of groups. For all subsequent calculations and filtering, raw data were re-analysed.

PCA scores plot of all samples and QCs filtered based on 90 % presence in at least one of two groups. Groups of samples are circled to show each group of samples and QCs. Cumulative R 2 and Q 2 were calculated to be 0.313 and 0.107, respectively
3.2 QC RSD calculation
Different strategies for filtering based on QC were tested for QC pool data to elucidate the most reproducible method for this dataset. Data were filtered (i) based on at least 50 % presence and lower than 30 % RSD in respective QCs, where missing values were included in RSD calculation; (ii) based on at least 50 % presence and lower than 30 % RSD in respective QCs, where missing values were not included in RSD calculation and (iii) based on at least 80 % presence and lower than 30 % RSD in respective QCs, where missing values were not included in RSD calculation. Filtering methods were compared through comparison of PCA models as shown in Fig. 3.

PCA scores plots of different filtering strategies based on QC samples. Filtering based on 50 % presence was performed without (missing value = NaN) and with (missing value = 0) considering missing values in CV calculation. Cumulative R 2 and Q 2 for these models were 0.286 and 0.061 and 0.445 and 0.284, respectively. For data filtered by 80 % presence in QCs, missing values were not considered in the calculation of CV. Cumulative R 2 and Q 2 for this model were calculated as 0.349 and 0.153. Filled circles Group A, open circles Group B, grey triangles QC
As can be seen in the PCA scores plots, QCs were best clustered following filtering based on 50 % presence in QCs, considering missing values. However, on interpretation of the data, it was observed that this filtering method resulted in the lowest number of features in the final dataset (757), as opposed to the other method of filtering based on 50 % presence (1,884) and the method based on 80 % (1,070). The reproducibility of QC data may not reflect that of the samples. For this reason, the most stringent filtering method could lead to loss of information that would have been relevant when comparing biological groups.
The source of missing values in metabolomics data is usually associated with analytical error (usually a higher number of missing values) or data reprocessing errors (usually a lower number of missing values). In theory, filtering based on 80 % presence in QCs retains features with lower numbers of missing values, attributed to reprocessing errors; therefore this option best reflects the ‘real’ data. This method offers the best compromise since it requires detection in a higher frequency of QC samples, but does not penalise features that had a very low number of missing values that could lead to their expulsion from the dataset in RSD calculation.
Data were subsequently filtered based on 90 % presence (in this case 7/8 samples—87.5 % which was rounded to 90 %) in at least one of the two groups and missing values in data were replaced as described in Sect. 2.
3.3 Comparison of QCs
Figure 4 shows the PCA plots for data filtered by each different QC. QCs clustered well in each case and the positioning of QCs in the plot was concordant with the types of QC displayed in each. A slight spread in QC surrogate was observed, however, since there was no observed trend due to analysis (Fig. S4, SI), this spread could not be explained by variation due to the order of measurement. As would be expected, the positions of the QCs in the separately prepared QCs (QCs A and B) were close to their respective groups.

PCA plots of samples filtered based on at least 80 % presence and lower than 30 % coefficient of variance in respective QCs. Cumulative R 2 and Q 2 were calculated as 0.405 and 0.193 for QC pool, 0.387 and 0.196 for QC surrogate, 0.344 and 0.185 for QC A and 0.488 and 0.362 for QC B. Filled circles Group A, open circles Group B, grey triangles QC
During the analysis of data at the chromatogram level, it was observed that the signal of spiked metabolites was reduced in QC pool samples and that QCs A and B were closer to their respective groups (Fig. S5, SI). For this reason, using QCs A and B could be advantageous over QC pool since true biomarkers could be lost in QC filtering with QC pool. Moreover, the total useful signal (Godzien et al. 2010) in QC pool samples (signal corresponding to relevant data) was lower than QCs A and B (8.21 × 107 for QC pool compared to 8.29 × 107 and 8.33 × 107 for QCs A and B respectively). The advantages of this are twofold: higher total useful signal could be indicative of signal from a greater number of features (higher metabolite coverage), and enhanced signal in separate QCs could be due to the lack of dilution effect as occurs in QC pool. Figure 5 illustrates the number of features after QAopt performed for each QC separately. As can be seen, the total number of features when summing information from QCs A and B was higher than QC pool by more than 300 features.

Venn diagram of the number of features following QAopt procedure performed for each type of QC separately
Analysing the data with respect to each spiked metabolite in different QCs revealed different levels of information about the QCs. Table 2 shows the % RSD calculated for each spiked metabolite in the different groups of QC.
The first clear observation is that three of the metabolites would be filtered out of the analysis if QC surrogate was used due to absence, or presence with unacceptable % RSD if the traditional method of filtering was applied (Dunn et al. 2011). This highlights the necessity to revise filtering options when working with QC surrogate (since it is assumed that selecting another type of QC cannot be an option due to experimental design).
Second, although the intensity of each spiked metabolite was higher in QCs A and B compared to the pool (Fig. S5, SI), the % RSD was not always lower. This demonstrates that reproducibility cannot always be improved with intensity. Furthermore, differences between the numbers of features with similar % RSD were not different between QCs A, B and QC pool (Fig. S6, SI). This highlights that although the total useful signal was higher in QCs A and B than pool, there was negligible difference on the reproducibility of the data. This was unexpected since splitting QCs into two samples was theorised to improve matrix and dilution effects and subsequently reproducibility.
Irrespective of the samples, the QCs analysed and the way they are employed in filtering and data treatment changes the dataset. The result of this can be the decision to keep or filter out real biomarkers. It is clear that QC surrogate is expected to be different to the samples, and therefore care must be taken in filtering. However, as can be seen in Fig. 6, QC pool samples are different to samples, which is a fact often overlooked. This highlights the importance of such an investigation to reveal the best options to work with different QCs. The lowest number of features different between QCs and samples occurred for QCs A and B which further proves that these QCs are the most related to the samples and potentially offer the best option in an experimental design. Furthermore, if QC pool or QC surrogate have to be selected, different strategies must be employed to account for the differences between them and samples. Although the numbers of features different between samples and QCs seem to be high, it should be clarified that the number does not reflect features present only in QCs (absent in samples), it reflects the number of features that did not pass filters either in QCs or samples.

Venn diagrams of the overlap between samples and different types of QCs after filtering with respective QC. Groups were filtered based on 90 % presence in its respective group and QCs were filtered based on 80 % presence and lower than 30 % RSD
In addition to QAopt, different filtering options were introduced to observe their effects on statistical outcomes. This could be measured by comparing the consequence of different filtration strategies for different types of QC on the rate of true/false positives from statistical analysis.
QA+ was introduced as a solution to recover information lost in QC filtering. The reproducibility of features that are not present in QCs cannot be assed, although they could be key biomarkers in the real samples. QA+ is based on QAopt, with the addition of including features absent in QCs and present in a high proportion of samples from one of the groups. Features were added to QA filtered data when their presence was high enough to pass the filtration based on groups, even though they were absent in QCs. Simultaneous filtration based on QCs and groups allowed the easy identification of such variables. QA+ was expected to be particularly useful for both QC pool (where features can be lost by dilution effect) and QC surrogate analyses. For these datasets, 18 and 32 features were added to QC pool and QC surrogate, respectively. The success of this method was measured by comparing the rate of true/false positives from statistical analysis.
Information from QC filtering is also commonly lost in the case where features are only detected in one group and are completely absent in the other and are therefore often absent from QC samples (Ciborowski et al. 2012a). To reflect this situation, additional in silico features were altered in the data through deleting values in one group while maintaining the original values for the other group in a random selection of eight features. Four features were altered to be present only in group A samples and four only in group B. Values for QCs were altered such that they (i) were present or absent according to their respective group for QCs A and B, (ii) were absent in all QC surrogate samples since case specific metabolites would not be expected to be present in external plasma and (iii) were altered for QC pool such that some features would be missed as metabolites present only in one group can be diluted and not observed due to their origin in the samples. Table 3 details the formation of in silico altered metabolites. A different number of features were altered to be present in QC pool in order to highlight the effect of different filtering strategies in different scenarios.
The effect of different filtration strategies on the statistical outcome was compared for all spiked and in silico altered metabolites. Table 4 highlights the results for each type of QC. Equally for QC pool and QC surrogate, both QA and QA+ were tested. Additionally, not filtering QC surrogate data was compared to observe its effect on the balance between true and false positives. Both metabolites spiked at equal concentrations were found to be true negatives.
All spiked metabolites added at varying concentrations were found to be statistically significant from QAopt and QA+ for QC pool and QCs A and B methods, however it was possible only through using QCs A and B to obtain all spiked and in silico added metabolites as true positives. Although from the analysis of spiked metabolites there was not a significant difference between QAopt and QA+ methods, in the case that features of interest are present only in one group and not the other (as tested by the in silico addition of features), QA+ improves the true positive rate by 50 % without affecting the false positive rate. Additionally, for in silico added metabolites, the same improvement could be made for QC surrogate working with no filtration compared to QA+. It can also be observed from these data that no filtration offers the best solution working with QC surrogate.
One important consideration to take into account is the goal of the study. For biomarker discovery, maintaining a lower level of false positives is more important than improving the true positive rate. On the other hand, in pathophysiological exploration studies, a higher true positive rate is desired regardless of the false positive rate. For these reasons the data in the table can be interpreted in different ways. For example, if the goal of a study utilising QC surrogate whereby data contain features present exclusively in one group, the best filtration for biomarker discovery would be QAopt, however for understanding the mechanism of a condition, no filtration offers 100 % true positive rate.
As the observed false positive rate was high regardless of the type of QC and strategy for data treatment, recursive analysis was employed in an attempt to minimise false positives due to artefacts in data pre-treatment. Table 5 shows the same results following recursive analysis of significant features. In all cases, recursive analysis reduced the false positive rate without affecting the true positive rate. Specifically, it can be seen that working with recursive analysis in conjunction with QC surrogate, whether or not metabolites are present in both groups or exclusively in one, no filtration offers the best solution. Additionally, recursive analysis greatly improves the results working with QCs A and B.
The fact that no filtration provided the best option for QC surrogate was likely due to the true positives being strong markers. In this way, true positives were not lost through QC filtration and false positives were reduced due to a significantly larger number of features being tested in the Bonferroni false positive rate test (~25 % more). However if the biomarkers were not strong markers, there is a risk of losing them with no filtration through Bonferroni false positive rate testing, therefore, QA+ can offer a reasonable compromise to working with QC surrogate.
3.4 Creation of a second dataset
Surrogate QC samples were introduced in metabolomics studies as an option to analyse batches of samples from a large cohort (Dunn et al. 2011). Although QA filters were proposed as the method to deal with such data, it could also be more appropriate, as shown in the present study, to follow no filtration based on QCs. Working with no filtration of QCs, it is not possible to perform batch correction in large studies. Therefore, in order to justify the possibility to work with no filtration, another dataset was created to show the effects of using (not-filtered) batches separately through validating one with another.
A second dataset was created from the acquired data whereby a scaling factor was applied to each sample to simulate another batch. Reviewing several datasets, it was observed that the variation in total signal between samples is on average 0.2 and for QCs 0.05. Therefore, a scaling factor was randomly created for each based on these tolerances. The second dataset was used both in batch correction by LOESS (Dunn et al. 2011) using an in-house written script for MATLAB and for validation of the first batch.
Although batches cannot be merged as described previously, if the experimental design permits (there are enough samples per experimental group in order to perform robust statistical analysis), batches can be used separately with the option of validating one with the other—performing statistics separately for each dataset and comparing final results.
Two strategies for combining data were tested: (i) two batches (acquired and created datasets) were treated separately following exactly the same data treatment (described in Sect. 2); (ii) two batches were combined together and signal correction through LOESS was applied before statistical analysis performed in the same way as described previously. To compare the obtained results, lists of p-values obtained for the acquired data was compared both with the created data and the LOESS batch-matched data as a reference. Comparing LOESS batch matched data and data whereby one batch was used to validate the other, there was no effect on the true positive rate, however the false positive rate was reduced by ~50 % (Fig. 7).

Venn diagrams of statistically significant masses between a acquired data and created data treated separately for statistical analysis and b between the acquired data and the results after batch matching both datasets through LOESS
As shown form this particular investigation, combination of datasets in large scale studies is still possible, even without filtering based on QCs and following the LOESS procedure. Depending on the experimental design this could offer an attractive alternative for studies with QC surrogate.
4 Conclusions
To elucidate the effect of different QCs on the analysis and final interpretation of data, a controlled experiment has been designed. Choices made in data treatment can have a considerable impact on the final dataset and on the results of statistical analyses. The presented data portray that the most relevant type of QC (if the experimental design permits) is the use of separate pools of QCs for each experimental group (QCs A and B). If it is not possible to do this (due to a very limited sample volume), and either QC pool or QC surrogate is necessary then the way they are used in data treatment should be carefully planned. From the results presented, the best way to deal with QC surrogate is to work without any filters based on QC (filtering should be only group specific). Using QC pool, QA+ offers a suitable option whereby features can be ‘saved’ and the true positive rate can be improved without affecting the false positives. Finally, recursive analysis has been suggested as a valuable method to recover data that ultimately improves statistical analyses (true positives are increased further with no effect on the false positives).
References
Ciborowski, M., Lipska, A., Godzien, J., Ferrarini, A., Korsak, J., Radziwon, P., et al. (2012a). Combination of LC–MS- and GC–MS-based metabolomics to study the effect of ozonated autohemotherapy on human blood. Journal of Proteome Research, 11, 6231–6241. doi:10.1021/pr3008946.
Ciborowski, M., Teul, J., Martin-Ventura, J. L., Egido, J., & Barbas, C. (2012b). Metabolomics with LC–QTOF–MS permits the prediction of disease stage in aortic abdominal aneurysm based on plasma metabolic fingerprint. PLoS ONE, 7, e31982. doi:10.1371/journal.pone.0031982.
Ciborowski, M., Ruperez, J., Martinez-Alcazar, M. P., Angulo, S., Radziwon, P., Olszanski, R., et al. (2010). Metabolomic approach with LC–MS reveals significant effect of pressure on diver’s plasma. Journal of Proteome Research, 9, 4131–4137. doi:10.1021/pr100331j.
Dunn, W. B., Broadhurst, D., Begley, P., Zelena, E., Francis-McIntyre, S., Anderson, N., et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols, 6, 1060–1083. doi:10.1038/nprot.2011.335.
Gika, H. G., Theodoridis, G. A., Earll, M., & Wilson, I. D. (2012). A QC approach to the determination of day-to-day reproducibility and robustness of LC–MS methods for global metabolite profiling in metabonomics/metabolomics. Bioanalysis, 4, 2239–2247. doi:10.4155/bio.12.212.
Gika, H. G., Theodoridis, G. A., Wingate, J. E., & Wilson, I. D. (2007). Within-day reproducibility of an HPLC–MS-based method for metabonomic analysis: Application to human urine. Journal of Proteome Research, 6, 3291–3303. doi:10.1021/pr070183p.
Godzien, J., Ciborowski, M., Angulo, S., & Barbas, C. (2013a). From numbers to a biological sense: How the strategy chosen for metabolomics data treatment may affect final results. A practical example based on urine fingerprints obtained by LC–MS. Electrophoresis, 34, 2812–2826. doi:10.1002/elps.201300053.
Godzien, J., Ciborowski, M., Whiley, L., Legido-Quigley, C., Ruperez, F. J., Barbas, C., et al. (2013b). In-vial dual extraction liquid chromatography coupled to mass spectrometry applied to streptozotocin-treated diabetic rats. Tips and pitfalls of the method. Journal of Chromatography A, 1304, 52–60. doi:10.1016/j.chroma.2013.07.029.
Godzien, J., et al. (2010). Metabolomic approach with LC-QTOF to study the effect of a nutraceutical treatment on urine of diabetic rats. Journal of Proteome Research, 10, 837–844. doi:10.1021/pr100993x.
Guy, P. A., Tavazzi, I., Bruce, S. J., Ramadan, Z., & Kochhar, S. (2008). Global metabolic profiling analysis on human urine by UPLC-TOFMS: Issues and method validation in nutritional metabolomics. Journal of Chromatography B, 871, 253–260. doi:10.1016/j.jchromb.2008.04.034.
Kamleh, M. A., Ebbels, T. M. D., Spagou, K., Masson, P., & Want, E. J. (2012). Optimizing the use of quality control samples for signal drift correction in large-scale urine metabolic profiling studies. Analytical Chemistry, 84, 2670–2677. doi:10.1021/ac202733q.
Llorach, R., Urpi-Sarda, M., Jauregui, O., Monagas, M., & Andres-Lacueva, C. (2009). An LC–MS-based metabolomics approach for exploring urinary metabolome modifications after cocoa consumption. Journal of Proteome Research, 8, 5060–5068. doi:10.1021/pr900470a.
Sangster, T., Major, H., Plumb, R., Wilson, A. J., & Wilson, I. D. (2006). A pragmatic and readily implemented quality control strategy for HPLC–MS and GC–MS-based metabonomic analysis. Analyst, 131, 1075–1078. doi:10.1039/b604498k.
Acknowledgments
Authors would like to acknowledge funding from the Ministry of Science and Technology (MCIT CTQ2011-23562).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Godzien, J., Alonso-Herranz, V., Barbas, C. et al. Controlling the quality of metabolomics data: new strategies to get the best out of the QC sample. Metabolomics 11, 518–528 (2015). https://doi.org/10.1007/s11306-014-0712-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-014-0712-4