
Abstract
Glial cells are involved in multiple cerebral functions that profoundly influence brain tissue viability during ischemia, and astrocytes are the main source of extracellular purines as adenosine and guanosine. The endogenous guanine-based nucleoside guanosine is a neuromodulator implicated in important processes in the brain, such as modulation of glutamatergic transmission and protection against oxidative and inflammatory damage. We evaluated if the neuroprotective effect of guanosine is also observed in cultured cortical astrocytes subjected to oxygen/glucose deprivation (OGD) and reoxygenation. We also assessed the involvement of A1 and A2A adenosine receptors and phosphatidylinositol-3 kinase (PI3K), MAPK, and protein kinase C (PKC) signaling pathways on the guanosine effects. OGD/reoxygenation decreased cell viability and glutamate uptake and increased reactive oxygen species (ROS) production in cultured astrocytes. Guanosine treatment prevented these OGD-induced damaging effects. Dipropyl-cyclopentyl-xanthine (an adenosine A1 receptor antagonist) and 4-[2-[[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl] benzenepropanoic acid hydrochloride (an adenosine A2A receptor agonist) abolished guanosine-induced protective effects on ROS production, glutamate uptake, and cell viability. The PI3K pathway inhibitor 2-morpholin-4-yl-8-phenylchromen-4-one, the extracellular-signal regulated kinase kinase (MEK) inhibitor 2′-amino-3′-methoxyflavone, or the PKC inhibitor chelerythrine abolished the guanosine effect of preventing OGD-induced cells viability reduction. PI3K inhibition partially prevented the guanosine effect of reducing ROS production, whereas MEK and PKC inhibitions prevented the guanosine effect of restoring glutamate uptake. The total immunocontent of the main astrocytic glutamate transporter glutamate transporter-1 (GLT-1) was not altered by OGD and guanosine. However, MEK and PKC inhibitions also abolished the guanosine effect of increasing cell-surface expression of GLT-1 in astrocytes subjected to OGD. Then, guanosine prevents oxidative damage and stimulates astrocytic glutamate uptake during ischemic events via adenosine A1 and A2A receptors and modulation of survival signaling pathways, contributing to microenvironment homeostasis that culminates in neuroprotection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemia is an interruption of blood supply to the tissue which results in inadequate oxygenation and glucose delivery. During ischemia, there is a progressive failure of cellular ATP production that impairs the activity of ionic pumps and causes disruption of the ionic gradient, causing release of excitatory amino acids such as glutamate [1]. This reversal activity of glutamate transporters, causing the leakage of glutamate and sodium (Na+) ions, also promotes failure of glutamate uptake from extracellular space and contributes to excitotoxicity [2].
Astrocytes are involved in a number of functions related to maintenance of neural cells viability during ischemia, such as water balance, blood–brain barrier integrity, cerebral blood flow regulation, ionic balance, and glutamate homeostasis [3, 4]. Astrocytic glutamate uptake is a crucial process for maintaining extracellular glutamate levels at physiological concentrations, thus preventing excitotoxicity [5, 6]. This process is achieved through specific high-affinity Na+-dependent excitatory amino acid transporters (EAATs) present in high concentration in astrocytes membrane. Astrocytic EAATs GLAST and glutamate transporter-1 (GLT-1) are directly related to regulation of extracellular glutamate levels and to neuroprotective effects [7]. GLT-1 alone is responsible for > 90% of total glutamate uptake [8]. EAATs remove extracellular glutamate through plasma membrane using the electrochemical Na+ and potassium (K+) gradients as the driving force, and nearly 90% of the glucose demand in the brain is used to maintain the electrochemical gradients necessary for glutamate uptake and neuronal depolarization [9]. Additionally, impairment in glutamate transport could be the result of intracellular reactive oxygen species (ROS) generation during ischemia and reperfusion, as glutamate transporters are vulnerable to oxidation due to the presence of thiol-based redox regulatory sites, thus resulting in decreased glutamate uptake [10]. The failure in energy production in brain ischemia compromises the activity of glutamate transporters and contributes to excitotoxic cell death. Therefore, astrocytes represent potential targets from a protective therapeutic perspective [2].
Astrocytes are also the major source of extracellular purinergic nucleosides guanosine and adenosine. The release of these nucleosides is stimulated under brain damaging conditions [11]. The local concentration of guanosine remained elevated for several days after a focal cerebral ischemia in rats [12]. Cultured astrocytes subjected to hypoxia/hypoglycemia release guanosine, and its extracellular levels continued to increase while accumulation of adenosine metabolites indicated its rapid metabolization [13]. Several studies demonstrated that guanosine presents neurotrophic and neuroprotective effects. Regarding neuroprotection, guanosine can modulate glutamatergic transmission, thus decreasing excitotoxicity (for review, see [14]). Studies from our research group demonstrated the protective effect of guanosine during an oxygen/glucose deprivation (OGD) insult in hippocampal slices. Guanosine inhibits the decrease in cell viability and the increase in ROS production induced by OGD [15,16,17]. OGD also decreased glutamate uptake and increased glutamate release, and those effects were similarly prevented by guanosine. Interestingly, the pharmacological blockade of the glutamate transporter GLT-1 inhibited guanosine effect of reducing glutamate release and cell damage induced by OGD [18].
We also demonstrated the participation of adenosine receptors and intracellular protein kinases related to cell viability maintenance in the neuroprotective effect of guanosine [19]. In hippocampal slices subjected to OGD, we observed that an adenosine A1 receptor (A1R) antagonist and an adenosine A2A receptor (A2AR) agonist inhibited the protective effect of guanosine. It is interesting to highlight that an A2AR antagonist did not alter the guanosine effect, pointing to an opposite effect of guanosine on the modulation of these receptors. Moreover, inhibition of the phosphatidylinositol-3 kinase (PI3K) or the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK) abolished the guanosine prevention of ROS production. Guanosine-induced recovery of glutamate uptake following OGD also involved activation of MAPK/ERK [16]. Therefore, we previously demonstrated that guanosine is protective against ischemia in a brain slice model [16, 18]. However, it is not clear whether all cell types within brain slices share similar molecular targets involved in the mechanism of guanosine-induced protective effects.
Considering that (i) astrocytes are the main source of extracellular nucleosides, (ii) they express P1 and P2 purinergic receptor subtypes for adenine-based purines, (iii) they act in conditions of brain repair [11], and (iv) that guanosine modulates glutamate transport via transporters expressed in astrocytes, this study investigated the protective role of guanosine against OGD in cortical cultured astrocytes. We also evaluated glutamate transporter activity and expression, as well as the involvement of adenosine A1R and A2AR, the intracellular signaling pathways PI3K and MAPK/ERK, and protein kinase C (PKC) pathways in the protective mechanism induced by guanosine.
Materials and methods
Animals
All experiments were performed in accordance with the “Principles of laboratory animal care” (NIH 2011) and were approved by the local Ethics Committee on the Use of Animal to Research (CEUA/UFSC-PP00955). Newborn (0–2-day-old) Wistar rats (male and female) were provided by the animal facility of the Universidade Federal de Santa Catarina (UFSC, Florianópolis, Brazil). Rat dams were kept in controlled conditions at 23 ± 1 °C, under a 12/12-h light–dark cycle (lights on at 7 am), with free access to water and food.
Cell culture
Primary cultures of astrocytes were prepared from cerebral cortex of newborn (0–2-day-old) Wistar rats as previously described [20]. Briefly, astrocytes were plated into 24-well plates (3.5 × 105cells) and grown to confluence (8–14 days) in the presence of Dulbecco’s modified Eagle’s medium and nutrient mixture F12 (DMEM-F12; Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS; Cultilab, SP). Cultures were incubated at 37 °C in a humidified 5% CO2 and 95% atmosphere.
Astrocyte treatment
Oxygen/glucose deprivation (OGD) in cultured astrocytes was performed by replacing the culture medium by an oxygen/glucose deprived buffer (ischemic buffer) in Hank’s balanced salt solution (HBSS, composition in mM: 1.3 CaCl2, 137 NaCl, 5 KCl, 0.65 MgSO4, 0.3 Na2HPO4, 1.1 KH2PO4 and, 5 HEPES, where 10 mM D-glucose was replaced by 10 mM 2-deoxy-glucose) [21]. Cultured astrocytes were then incubated in an anaerobic chamber containing a gas mixture of 95% N2/5% CO2 for 150 min [22]. After this OGD period, cultured astrocytes were incubated with standard culture medium and maintained in a 5% CO2 humidified atmosphere at 37 °C for 24 h of reoxygenation.
Guanosine was added to the control group (GUO group) or during reoxygenation (OGD GUO group). Adenosine receptor ligands, dipropyl-cyclopentyl-xanthine (DPCPX) (100 nM, A1R antagonist) and 4-[2-[[6-amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl] benzenepropanoic acid hydrochloride (CGS21680) (200 nM, A2AR agonist), and protein kinase inhibitors, chelerythrine (1 μM, PKC inhibitor, Sigma), 2-morpholin-4-yl-8-phenylchromen-4-one (LY294002) (10 μM, PI3K inhibitor, Sigma), and 2′-amino-3′-methoxyflavone (PD98059) (10 μM, MEK inhibitor, Sigma), were used to assess the targets and signaling pathways involved in neuroprotection promoted by guanosine. Adenosine receptor ligands or protein kinase inhibitors were pre-incubated for 15 min before adding guanosine (GUO) at the beginning of reoxygenation period, and they were kept together during 24 h of incubation. A schematic overview of the protocol treatment is depicted at Fig. 1.

Guanosine protects against cell damage induced by OGD in cortical astrocytic cells. A schematic overview of the treatment protocol is represented in the top of the figure. a Astrocytic cells were incubated with increasing concentrations of guanosine (GUO) 10, 50, 100, or 500 μM in physiological culture medium for 24 h, and cell viability was measured by MTT reduction. b In vitro model of ischemia: astrocytic cells were subject to 2.5 h of oxygen and glucose deprivation (OGD) followed by 24 h of reoxygenation with culture medium (control) or with increasing concentrations of guanosine (OGD GUO, 10–500 μM). Evaluation of adenosine receptors and signaling pathways involvement: c after 2.5 h OGD in the beginning of reoxygenation period, astrocytes were pre-incubated for 30 min with DPCPX (100 nM, A1R antagonist); d with CGS21680 (CGS, 200 nM, A2AR agonist); e LY294002 (LY, 10 μM, a PI3K inhibitor), or PD98059 (PD, 10 μM, MEK/ERK inhibitor), or chelerythrine (CHE, 1 μM, a PKC inhibitor), and subsequently co-incubated 24 h with guanosine (GUO, 10 μM) in culture medium. Cellular viability was evaluated by MTT reduction assay and was expressed as percentage of control cells representing cells incubated for 24 h in culture medium (100% of cellular viability). Data are means ± S.E.M. from 4 to 5 different cell batches. *p < 0.05 represents means significantly different from Control and OGD + GUO groups. #p < 0.05 represents means significantly different from OGD group (ANOVA followed by Tukey’s test)
MTT reduction
Cell viability was determined through the ability of cells to reduce MTT (3-(4,5-dimethylthiazol-2-yl-diphenyltetrazolium bromide; Sigma) [23]. After 24 h of reoxygenation, cultured astrocytes were incubated with MTT (0.5 mg/ml) in PBS for 2 h at 37 °C. The tetrazolium ring of MTT can be cleaved by active dehydrogenases in order to produce a precipitated formazan. The medium was withdrawn, the precipitated formazan was solubilized with dimethyl sulfoxide, and viable cells were quantified spectrophotometrically at a wavelength of 550 nm.
Measurement of ROS production
In order to measure cellular ROS production, we used the molecular fluorescent probe H2DCFDA. After the 24 h period of reoxygenation, astrocytes were loaded with 10 μM H2DCFDA for 20 min. H2DCFDA diffuses through the cell membrane and is hydrolyzed by intracellular esterases to the non-fluorescent form 2′,7′-dichlorodihydrofluorescein (DCFH). DCFH reacts with intracellular H2O2 to form 2′,7′-dichlorofluorescein (DCF), a green fluorescent dye. Fluorescence was measured in a fluorescence microplate reader (TECAN). Wavelengths of excitation and emission were 485 and 520 nm, respectively [17].
l-[3H] glutamate uptake
l-[3H]glutamate uptake was evaluated as previously described [24]. After OGD and 24 h reoxygenation protocol, astrocytic cells were washed for 15 min at 37 °C in HBSS, composition in mM: 1.3 CaCl2, 137 NaCl, 5 KCl, 0.65 MgSO4, 0.3 Na2HPO4, 1.1 KH2PO4, 2 glucose, and 5 HEPES. Uptake was assessed by adding 0.33 μCi/ml l-[3H]glutamate with 100 μM unlabeled glutamate in a final volume of 300 μl. Incubation was stopped immediately after 7 min by discarding the incubation medium, and astrocyte cells were subjected to two ice-cold washes with 1 ml HBSS. Astrocyte cells were solubilized by adding a solution with 0.1% NaOH/0.01% SDS and incubated overnight. Aliquots of slice lysates were taken for determination of the intracellular content of l-[3H] glutamate by scintillation counting. Sodium-independent uptake was determined by using choline chloride, instead of sodium chloride in the HBSS buffer. Unspecific sodium-independent uptake was subtracted from total uptake to obtain the specific sodium-dependent glutamate uptake. Results were expressed in nanomoles of l-[3H] glutamate and taken up per milligram of protein per minute.
Western blot analysis
The following OGD and guanosine treatment astrocytes were washed once with cold phosphate-buffered saline (PBS/EDTA), harvested and lysed by mechanical homogenization in 100 μl of ice-cold lysis buffer (Tris 50 mM pH 7.0, EDTA 1 mM, NaF 100 mM, phenylmethylsulfonyl fluoride (PMSF) 0.1 mM, Na3VO4 2 mM, Triton X-100 1%, glycerol 10% and Sigma Protease Inhibitor Cocktail (P2714)). Lysates were centrifuged (10,000×g for 10 min, at 4 °C) to eliminate cellular debris. The supernatants were diluted 1/1 (v/v) in Tris 100 mM pH 6.8, EDTA 4 mM, SDS 8% and boiled for 5 min. After that, samples were diluted (40% glicerol, 100 mM Tris, bromophenol blue, pH 6.8) in the ratio 25:100 (v/v) and β mercaptoethanol (final concentration 8%). The same amount of protein (60 μg per lane) for each sample was separated by SDS–PAGE in 10% minigels and transferred to nitrocellulose membranes by using a semidry blotting apparatus (1.2 mA/cm2; 1.5 h). Membranes were blocked with 5% skim milk in Tris-buffered saline (TBS, Tris 10 mM, NaCl 150 mM, pH 7.5). GLT-1 was detected after overnight incubation with specific antibody diluted in TBS-T (Tris 10 mM, NaCl 150 mM, Tween-20 0.1%, pH 7.5) containing bovine serum albumin (BSA) 2% in the dilution of 1:1000 (anti-GLT-1, Santa Cruz®; rabbit). Membranes were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (1:5000) for GLT-1 detection. The reactions were developed by chemiluminescence substrate (LumiGLO). All blocking and incubation steps were followed by three steps of washing for 5 min with TBS-T. All membranes were incubated with mouse anti-β-actin (Cell Signaling®; 1:1000) antibody to verify the protein loading of gels. The immunocontent was determined as a ratio of optical density (OD) of GLT-1 band/OD of the β-actin band [25, 26]. Bands were quantified by using the Scion Image® software.
Immunocytochemistry
Astrocyte monolayers cultured on poly-l-lysine-treated (0.1 mg/mL) coverslips were fixed with 4% paraformaldehyde during 30 min. In order to perform cells surface GLT-1 immunodetection, non-permeabilized cells were incubated with the polyclonal antibody anti-GLT-1 (1:500, Santa Cruz; rabbit) overnight (at 4 °C), followed by incubation with secondary antibody to rabbit IgG Alexa Fluor® 488 (1:1.000, Millipore; at room temperature, for 1 h). Nuclear staining was performed by incubating cells with Hoechst33342 (5 μg/mL) in PBS for 10 min at room temperature and then washed twice with PBS (pH 7.4). Cultures were air dried and mounted onto glass slides, which were examined under a confocal microscope (Leica DMI6000 B). Three independent experiments assayed in triplicate were analyzed to quantify GLT-1 intensity of fluorescence using the LAS-AF program version 2.6.0 (Leica Microsystem, Wetzlar, Germany). The intensity of fluorescence related to number of cells stained with Hoechst33342 was analyzed with ImageJ.
Protein measurement
Protein determination was evaluated by the method of Lowry [27] for glutamate uptake assay and by the method of Peterson [28] for western blotting analysis. Bovine serum albumin (Sigma) was used as standard.
Statistical analysis
For all protocols, values are means ± SEM (standard error of the mean) from 4 to 5 experiments from different cell batches and carried out in triplicate. Statistical analyses were performed by using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test with GraphPad software.
Results
We initially evaluated if guanosine per se could affect astrocytes viability by treating astrocyte cultures with increasing guanosine concentrations (10, 50, 100, or 500 μM) for 24 h. Guanosine treatment did not alter cellular viability of astrocytes maintained in physiological medium at any concentration tested (Fig. 1a).
To verify the protective effect of guanosine against OGD in cultured cortical astrocytes, cells were maintained for 150 min in an oxygen/glucose deprived (OGD) medium followed by 24 h of reoxygenation in standard culture conditions (OGD/reoxygenation protocol). Guanosine treatment occurred during the reoxygenation period. OGD (150 min) followed by reoxygenation (24 h) decreased astrocytic viability compared to control cells (Fig. 1b). Treatment with increasing guanosine concentrations (10–500 μM) during reoxygenation period promoted cellular protection at all tested concentrations (Fig. 1b). Subsequent experiments were performed with 10 μM guanosine, the lowest effective guanosine concentration observed in this study.
The evaluation of adenosine receptor involvement on guanosine effect was assessed by incubating cultured astrocytes with DPCPX (100 nM, an A1R antagonist) or CGS21680 (200 nM, an A2AR agonist) in the reoxygenation period. Neither DPCPX nor CGS21680 significantly altered the astrocytic viability when added in physiological conditions (Fig. 1c, d) or after the OGD/reperfusion period (data not shown). The incubation of DPCPX or CGS21680 before guanosine addition in the reoxygenation period abolished the guanosine protective effect on cultured astrocytes (Fig. 1c, d). These results reinforce the idea that adenosine receptors may be the molecular targets to guanosine-induced protection.
To access the involvement of signaling pathways related to the protective effect of guanosine in astrocytes, cells were incubated with selective protein kinases inhibitors (10 μM LY294002, a PI3K inhibitor; 10 μM PD98059, a MEK inhibitor; 1 μM chelerythrine, a PKC inhibitor) 15 min before guanosine treatment. As shown in Fig. 1e, inhibition of PI3K, MEK, or PKC pathways abolished the protective effect of guanosine against OGD.
Next, we evaluated the guanosine effect on ROS production, as oxidative stress is related to a decrease in cellular viability in cerebral ischemia. We observed an increase in ROS levels (65% higher than control group) in cultured astrocytes subjected to OGD/reoxygenation. Guanosine treatment abolished OGD-induced increase in ROS production (Fig. 2). Antagonism of A1R prevented the guanosine effect of reducing ROS production induced by OGD (Fig. 2a), whereas A2AR activation by its agonist CGS21680 partially prevented this guanosine effect (Fig. 2b).

Guanosine prevents ROS production elicited by OGD in cortical astrocytic cells. Astrocytes were subjected to 2.5 h of oxygen and glucose deprivation (OGD) followed by 24 h of reperfusion with culture medium (control) or with guanosine (OGD GUO, 10 μM). Evaluation of adenosine receptors and signaling pathways involvement: a at the beginning of reoxygenation period, astrocytes were pre-incubated for 30 min with DPCPX (100 nM, A1R antagonist); b with CGS21680 (CGS, 200 nM, A2AR agonist); c LY294002 (LY, 10 μM, a PI3K inhibitor); d PD98059 (PD, 10 μM, MEK/ERK inhibitor); e chelerythrine (CHE, 1 μM, a PKC inhibitor) and subsequently co-incubated 24 h with guanosine (GUO, 10 μM) in culture medium. ROS production was evaluated by DCF fluorescence emission assay and was expressed as fold increase from control (incubated for 24 h in physiological culture medium). Data are means ± S.E.M. from 4 to 5 different cell batches. *p < 0.05 represents means significantly different from Control and OGD + GUO groups. #p < 0.05 represents means significantly different from OGD group (ANOVA followed by Tukey’s test)
Evaluation of the signaling pathways involved in this guanosine effect showed that the PI3K inhibitor, LY294002, partially blocked the decrease of ROS production induced by guanosine (Fig. 2c). On the other hand, PD98059 (MEK inhibitor) or chelerythrine (PKC inhibitor) did not alter guanosine-induced decrease in ROS levels (Fig. 2d, e).
The effect of guanosine on glutamate uptake was evaluated as astrocytic glutamate transporters are the most effective in the clearance of extracellular glutamate. As expected, OGD dramatically reduces glutamate uptake in cultured astrocytes and guanosine treatment restored glutamate uptake to basal levels (Fig. 3a). Both A1R antagonist (DPCPX) and A2AR agonist (CGS21680) blocked guanosine-induced increase in glutamate uptake after OGD (Fig. 3a, b). The PI3K inhibitor, LY294002, did not alter guanosine-induced glutamate uptake stimulation (Fig. 3c). Interestingly, PD98059 and chelerythrine (MEK and PKC inhibitors, respectively) inhibited the guanosine effect of recovering glutamate uptake (Fig. 3d, e).

Guanosine prevents the decrease in glutamate uptake elicited by OGD in cortical astrocytic cells. Astrocytes were subject to 2.5 h of oxygen and glucose deprivation (OGD) followed by 24 h of reperfusion with culture medium (control) or with guanosine (OGD GUO, 10 μM). Evaluation of adenosine receptors and signaling pathways involvement: a in the beginning of reoxygenation period, astrocytes were pre-incubated for 30 min with DPCPX (100 nM, A1R antagonist); b with CGS21680 (CGS, 200 nM, A2AR agonist); c LY294002 (LY, 10 μM, a PI3K inhibitor); d PD98059 (PD, 10 μM, MEK/ERK inhibitor); e chelerythrine (CHE, 1 μM, a PKC inhibitor), and subsequently co-incubated 24 h with guanosine (GUO, 10 μM) in culture medium. Glutamate uptake was assessed by using l-[3H]glutamate, as described in “Materials and methods.” Data are means ± SEM from 5 different cell batches. *p < 0.05 represents means significantly different from Control and OGD + GUO groups. #p < 0.05 represents means significantly different from OGD group (ANOVA followed by Tukey’s test)
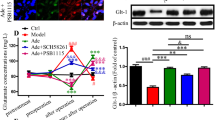
As glutamate transporter-1 (GLT-1) is ubiquitously expressed in the brain and it is the astrocytic glutamate transporter with higher glutamate uptake capacity [5], we performed GLT-1 immunodetection in cultured astrocytes after OGD/reoxygenation and guanosine treatment. Western blot analysis showed no alteration in the total immunocontent of GLT-1 in astrocyte homogenates following OGD and/or guanosine treatment (Fig. 4).

Immunodetection of GLT-1 in homogenates of astrocytes subjected to OGD/reoxygenation and treated with guanosine. Astrocytes were subject to 2.5 h of oxygen and glucose deprivation (OGD) followed by 24 h of reperfusion with culture medium (Control) or with guanosine (OGD GUO, 10 μM). Western blot analysis was performed as described in “Materials and methods.” Data are means ± SEM from 4 different cell batches assayed in triplicate. No statistical significance was observed (ANOVA)
Then, we decided to analyze the possible role of guanosine on the cellular distribution of GLT-1 in astrocytes subjected to ischemia. Trafficking of glutamate transporters to the plasma membrane and cell-surface expression is a crucial mechanism for regulating glutamate transporter function [29]. Immunocytochemistry analyses were carried out in non-permeabilized astrocyte cultures after OGD/reoxygenation and guanosine treatment. OGD promoted a 46% decrease in the levels of fluorescence labeling of cell-surface GLT-1 as compared to control group (Fig. 5a, b). Guanosine treatment prevented this decrease in cell-surface GLT-1 immunofluorescence in astrocytes, and the pre-incubation of PD98059 (OGD PD GUO) or chelerythrine (OGD CHE GUO) inhibited the effect of guanosine. This analysis indicates that guanosine restores GLT-1 expression in astrocytic cellular membranes following OGD/reoxygenation damage, and this effect depends on MAPK/ERK and PKC activation.

Immunocytochemical analysis of GLT-1 expression in non-permeabilized astrocytes subjected to OGD/reoxygenation and treated with guanosine. Astrocyte cells were subject to 2.5 h of oxygen and glucose deprivation (OGD) followed by 24 h of reperfusion with culture medium (Control) or with guanosine (OGD GUO, 10 μM). Cells were pre-incubated for 30 min with PD98059 (PD, 10 μM, MEK-ERK inhibitor), or chelerythrine (CHE, 1 μM, a PKC inhibitor) and subsequently co-incubated 24 h with guanosine (GUO). a Immunofluorescence for cell-surface GLT-1 labeling was performed as described in “Materials and methods.” Cell nuclei were stained with Hoechst33342. Images in a × 20 magnification are representative of three different cell batches assayed in triplicate. b The graphic shows means ± SEM of the immunofluorescence quantification showed as the relative intensity of fluorescence of GLT-1. *p < 0.05 represents means significantly different from Control and OGD + GUO groups (ANOVA followed by Tukey’s test)
Discussion
The maintenance of astrocytes viability and functionality after an ischemic event is fundamental for brain tissue recovery after injury. As guanosine affords cellular protection against glutamate excitotoxicity and displays trophic effects in glial cells [19, 30], we evaluated the possible protective effect of guanosine in astrocytes subjected to an in vitro ischemia protocol. We showed here that guanosine treatment decreases ROS production and increases cellular viability, glutamate uptake, and cell-surface expression of GLT-1 in cultured cortical astrocytes subjected to ischemia. Additionally, we showed the involvement of adenosine receptors and signaling pathways related to cell survival in the protective effect of guanosine in astrocytes.
Although neurons are more susceptible to ischemia, astrocytes also undergo damage after ischemic events. Astrocyte impairment contributes to the decrease of neuronal viability and functionality under ischemic conditions [31, 32]. The guanosine protective effect was also showed in other astrocytic cultures, as the C6 glial cell line [33, 34], in cultured hippocampal astrocytes subjected to lipopolysaccharide challenge [35], and in astrocytes obtained from aged rats [36]. In our study, guanosine treatment prevented OGD-induced reduction in cortical astrocytes viability. We previously showed the protective effect of guanosine in hippocampal slices subjected to an OGD protocol [15,16,17,18]. Therefore, we could hypothesize that guanosine promotes its neuroprotective effect by modulating astrocytes function, suggesting this cell type might be the cell target of guanosine in the brain. Interestingly, astrocytes are the main source of purines in the brain, mainly guanosine and adenosine, and astrocyte-mediated release of guanosine after an ischemic event was previously showed [13].
Ischemic events, particularly post-ischemic reperfusion, enhance ROS formation in brain tissue. Excessive ROS production may induce cell damage either directly, by interacting and compromising cellular protein integrity, lipids, and DNA, or indirectly, by affecting normal cellular signaling pathways and gene regulation [37]. Here we observed that OGD increased ROS levels in cultured astrocytes, which could be related to the decrease in astrocytes viability promoted by ischemia. We also showed that guanosine inhibited the increase in ROS levels induced by OGD in astrocytes. Guanosine antioxidant effects were showed in a glial cell line subjected to oxidative damage [33], in SH-5YSY neuroblastoma cells [25], in hippocampal slices subjected to OGD [16], and in animal models of ischemia [38, 39]. However, we recently showed that guanosine per se did not act as a ROS scavenger, but it prevents the production of ROS, nitric oxide, and peroxynitrite induced by OGD [17]. Therefore, guanosine may modulate protein targets and signaling pathways that prevent the oxidative burst, contributing to antioxidant defenses.
Investigation of drugs that reduce glutamate excitotoxicity through the maintenance of astrocytes functionality could be an interesting neuroprotective strategy against several neurological diseases where glutamate toxicity is a key element. Astrocytic glutamate transporters are essential for glutamatergic transmission fine-tuning, controlling glutamate uptake and release and preventing from excitotoxicity events. Evidence support the hypothesis that the neuroprotective effect of guanosine against excitotoxicity is related to the modulation of glutamate uptake [12, 16, 40,41,42]. In agreement, we confirmed that guanosine could act directly on astrocytes, by restoring the reduction of glutamate uptake and the reduction of cell-surface expression of GLT-1 induced by OGD/reoxygenation.
Our results provide additional information about guanosine mechanism of action. There is no specific molecular target (membrane receptor) identified to guanosine up to now [14, 43], although there is evidence for a putative G protein-coupled receptor selective to guanosine [44], to which is lacking molecular analysis. We recently reviewed the role for the well-characterized brain adenosine receptors in guanosine-induced biological effects, as neuroprotective and trophic effects [19]. In this regard, the A1R antagonist DPCPX and the A2AR agonist CGS21680 inhibited guanosine-induced neuroprotection in hippocampal slices subjected to OGD [16]. Similarly, in cultured astrocytes subjected to OGD, we observed that both DPCPX and CGS21680 abolished the decrease in ROS levels and the increase in cell viability and glutamate uptake promoted by guanosine. Interestingly, DPCPX had no effect over guanosine-induced glutamate uptake in hippocampal slices [16], suggesting that guanosine could modulate glutamatergic transporters activity independently of A1R in the presence of neuronal cells and that A1R activation in astrocytes is essential for guanosine-induced modulation of glutamate uptake.
Concerning the intracellular signaling pathways involved in the guanosine action, we showed that guanosine was effective in stimulating glutamate uptake after ischemic damage depending on MEK or PKC pathway activation in astrocytes. The involvement of MEK pathway in the modulation of astrocytic glutamate transporters had been demonstrated in previous studies [29, 45], as ERK1/2 activation is related to increased activity of glutamate transporters. In C6 glial cells [33] and in hippocampal slices [16], inhibition of PKC also counteracted guanosine-induced recovery of glutamate uptake after ischemia.
The involvement of PI3K/protein kinase B (AKT) pathway in guanosine modulation of glutamate uptake into astrocytes was also evaluated. PI3K/AKT pathway has been implicated in development, neuronal survival, and neuroplasticity [46, 47]. Activation of AKT is involved in the recruitment of proteins related to cell survival and inhibition of pro-apoptotic mediators [48]. The participation of PI3K/AKT pathway in neuroprotective effects has been shown to involve increased glutamate uptake [49]. Guanosine effects on astrocytic glutamate uptake showed here did not involve the PI3K/AKT activation. However, in a previous study performed in hippocampal slices subject to OGD, PI3K inhibition blocked the guanosine effect of increasing glutamate uptake [15]. This difference in signaling pathways involved in the effect of guanosine may be related to a cell type-specific mechanism of action of guanosine. It is possible that guanosine-induced glutamate uptake is mediated by PI3K in neurons, as excitatory amino acid carrier 1 (EAAC1) expression induced by trophic factor involves PI3K activation [50]. However, in cortical astrocytes subjected to OGD, PI3K/AKT pathway is not involved in guanosine-induced glutamate uptake.
Guanosine-induced increase in glutamate uptake may be related to its ability to improve glutamate transporters activity, increase their expression, or even alter the distribution of these transporters. Thus, we evaluated the expression of GLT-1 in astrocytes subjected to OGD and guanosine. No alteration was observed in the total immunocontent of GLT-1 following OGD/reoxygenation and guanosine treatment. However, we observed that OGD decreased the cell-surface GLT-1 immunofluorescence intensity as compared to control astrocytes. Guanosine treatment restored cell-surface expression of GLT-1 in astrocytes, and MEK or PKC inhibition abolished this effect. Induction of GLT-1 expression and cell membrane translocation of this transporter have been shown to involve ERK activation [51]. Guanosine effect was dependent on MAPK/ERK activation, although it was also dependent on PKC activation. Additional studies may unravel the organization of these signaling pathways regarding the modulation of subcellular distribution and function of GLT-1 disrupted by OGD and restored by guanosine. A schematic overview of guanosine glioprotective effects against ischemia is depicted at Fig. 6.

Schematic overview of guanosine-induced glioprotective effects against ischemia in cortical astrocytes. OGD/reoxygenation decreased cell viability, glutamate uptake and GLT-1 levels, and increased reactive oxygen species (ROS) production in cultured astrocytes. Guanosine treatment during the reoxygenation period (24 h) prevented these OGD-induced damaging effects. Adenosine A1R antagonist and A2AR agonist abolished guanosine-induced protective effects on ROS production, glutamate uptake, and cell viability. Inhibition of the signaling pathways PI3K, MEK, or PKC prevented the guanosine effect on cell survival. PI3K inhibition partially prevented the guanosine effect of reducing ROS production induced by OGD. MEK or PKC inhibition abolished the guanosine effects of restoring glutamate uptake and cell-surface GLT-1 levels in astrocytes subjected to OGD
In conclusion, guanosine promoted astrocytic cells protection by preventing the increase in ROS generation and restoring glutamate uptake and expression of GLT-1 in astrocytes membranes after an ischemic insult. The prevention of oxidative damage and stimulation of glutamate uptake induced by guanosine in astrocytes may contribute to the microenvironment homeostasis in an ischemic situation, thus affording neuroprotection. Guanosine effects of preventing gliotoxicity are dependent on adenosine A1R and A2AR modulation, and may recruit PI3K, and mainly MEK and PKC pathways in astrocytes.
Abbreviations
- A1R:
-
Adenosine A1 receptor
- A2AR:
-
Adenosine A2A receptor
- CGS21680:
-
4-[2-[[6-Amino-9-(N-ethyl-β-d-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl] benzenepropanoic acid hydrochloride
- DPCPX:
-
Dipropyl-cyclopentyl-xanthine
- ERK:
-
Extracellular signal-regulated kinase
- GLT-1:
-
Glutamate transporter-1
- GUO:
-
Guanosine
- HBSS:
-
Hank’s balanced salt solution
- KRB:
-
Krebs-Ringer bicarbonate buffer
- LY294002:
-
2-Morpholin-4-yl-8-phenylchromen-4-one
- MAPK:
-
Mitogen-activated protein kinase
- MEK:
-
Extracellular-signal regulated kinase kinase
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OGD:
-
Oxygen/glucose deprivation
- PD98059:
-
2′-Amino-3′-methoxyflavone
- PI3K:
-
Phosphatidylinositol-3 kinase
- PKC:
-
Protein kinase C
- ROS:
-
Reactive oxygen species
References
Bonde C, Sarup A, Schousboe A, Gegelashvili G, Zimmer J, Noraberg J (2003) Neurotoxic and neuroprotective effects of the glutamate transporter inhibitor DL-threo-beta-benzyloxyaspartate (DL-TBOA) during physiological and ischemia-like conditions. Neurochem Int 43(4–5):371–380
Camacho A, Massieu L (2006) Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res 37(1):11–18
Trendelenburg G, Dirnagl U (2005) Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia 50(4):307–320
Zhao Y, Rempe DA (2010) Targeting astrocytes for stroke therapy. Neurotherapeutics 7(4):439–451
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65(1):1–105
Schousboe A, Waagepetersen HS (2006) Glial modulation of GABAergic and glutamatergic neurotransmission. Curr Top Med Chem 6(10):929–934
Guillet BA et al (2005) Differential regulation by protein kinases of activity and cell surface expression of glutamate transporters in neuron-enriched cultures. Neurochem Int 46(4):337–346
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51(6–7):333–355
Dienel GA (2013) Astrocytic energetics during excitatory neurotransmission: what are contributions of glutamate oxidation and glycolysis? Neurochem Int 63(4):244–258
Trotti D, Danbolt NC, Volterra A (1998) Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci 19(8):328–334
Ciccarelli R, Ballerini P, Sabatino G, Rathbone MP, D’Onofrio M, Caciagli F, di Iorio P (2001) Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci 19(4):395–414
Uemura Y, Miller JM, Matson WR, Beal MF (1991) Neurochemical analysis of focal ischemia in rats. Stroke 22(12):1548–1553
Ciccarelli R, di Iorio P, Giuliani P, D’Alimonte I, Ballerini P, Caciagli F, Rathbone MP (1999) Rat cultured astrocytes release guanine-based purines in basal conditions and after hypoxia/hypoglycemia. Glia 25(1):93–98
Tasca CI, Lanznaster D, Oliveira KA, Fernández-Dueñas V, Ciruela F (2018) Neuromodulatory effects of guanine-based purines in health and disease. Front Cell Neurosci 12:376
Dal-Cim T, Martins WC, Santos ARS, Tasca CI (2011) Guanosine is neuroprotective against oxygen/glucose deprivation in hippocampal slices via large conductance Ca(2)+-activated K+ channels, phosphatidilinositol-3 kinase/protein kinase B pathway activation and glutamate uptake. Neuroscience 183:212–220
Dal-Cim T, Ludka FK, Martins WC, Reginato C, Parada E, Egea J, López MG, Tasca CI (2013) Guanosine controls inflammatory pathways to afford neuroprotection of hippocampal slices under oxygen and glucose deprivation conditions. J Neurochem 126(4):437–450
Thomaz DT, Dal-Cim TA, Martins WC, Cunha MP, Lanznaster D, de Bem AF, Tasca CI (2016) Guanosine prevents nitroxidative stress and recovers mitochondrial membrane potential disruption in hippocampal slices subjected to oxygen/glucose deprivation. Purinergic Signal 12(4):707–718
Dal-Cim T, Martins WC, Thomaz DT, Coelho V, Poluceno GG, Lanznaster D, Vandresen-Filho S, Tasca CI (2016) Neuroprotection promoted by guanosine depends on glutamine synthetase and glutamate transporters activity in hippocampal slices subjected to oxygen/glucose deprivation. Neurotox Res 29(4):460–468
Lanznaster D, Dal-Cim T, Piermartiri TCB, Tasca CI (2016) Guanosine: a neuromodulator with therapeutic potential in brain disorders. Aging Dis 7(5):657–679
Mendes-de-Aguiar CB et al (2008) Thyroid hormone increases astrocytic glutamate uptake and protects astrocytes and neurons against glutamate toxicity. J Neurosci Res 86(14):3117–3125
Pocock JM, Nicholls DG (1998) Exocytotic and nonexocytotic modes of glutamate release from cultured cerebellar granule cells during chemical ischaemia. J Neurochem 70(2):806–813
Hurtado O, Lizasoain I, Fernández-Tomé P, Álvarez-Barrientos A, Leza JC, Lorenzo P, Moro MA (2002) TACE/ADAM17-TNF-alpha pathway in rat cortical cultures after exposure to oxygen-glucose deprivation or glutamate. J Cereb Blood Flow Metab 22(5):576–585
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65(1–2):55–63
Molz S, Dal-Cim T, Tasca CI (2009) Guanosine-5′-monophosphate induces cell death in rat hippocampal slices via ionotropic glutamate receptors activation and glutamate uptake inhibition. Neurochem Int 55(7):703–709
Dal-Cim T, Molz S, Egea J, Parada E, Romero A, Budni J, Martín de Saavedra MD, Barrio L, Tasca CI, López MG (2012) Guanosine protects human neuroblastoma SH-SY5Y cells against mitochondrial oxidative stress by inducing heme oxigenase-1 via PI3K/Akt/GSK-3beta pathway. Neurochem Int 61(3):397–404
Constantino LC, Binder LB, Vandresen-Filho S, Viola GG, Ludka FK, Lopes MW, Leal RB, Tasca CI (2018) Role of phosphatidylinositol-3 kinase pathway in NMDA preconditioning: different mechanisms for seizures and hippocampal neuronal degeneration induced by quinolinic acid. Neurotox Res 34(3):452–462
Lowry OH et al (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Peterson GL (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83(2):346–356
Gegelashvili G et al (2000) The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int 37(2–3):163–170
Decker H, Francisco SS, Mendes-de-Aguiar CBN, Romão LF, Boeck CR, Trentin AG, Moura-Neto V, Tasca CI (2007) Guanine derivatives modulate extracellular matrix proteins organization and improve neuron-astrocyte co-culture. J Neurosci Res 85(9):1943–1951
Lukaszevicz AC, Sampaïo N, Guégan C, Benchoua A, Couriaud C, Chevalier E, Sola B, Lacombe P, Onténiente B (2002) High sensitivity of protoplasmic cortical astroglia to focal ischemia. J Cereb Blood Flow Metab 22(3):289–298
Giffard RG, Swanson RA (2005) Ischemia-induced programmed cell death in astrocytes. Glia 50(4):299–306
Quincozes-Santos A, Bobermin LD, Souza DG, Bellaver B, Gonçalves CA, Souza DO (2014) Guanosine protects C6 astroglial cells against azide-induced oxidative damage: a putative role of heme oxygenase 1. J Neurochem 130(1):61–74
Giuliani P et al (2015) Guanosine protects glial cells against 6-hydroxydopamine toxicity. Adv Exp Med Biol 837:23–33
Bellaver B, Souza DG, Bobermin LD, Gonçalves CA, Souza DO, Quincozes-Santos A (2015) Guanosine inhibits LPS-induced pro-inflammatory response and oxidative stress in hippocampal astrocytes through the heme oxygenase-1 pathway. Purinergic Signal 11(4):571–580
Souza DG, Bellaver B, Bobermin LD, Souza DO, Quincozes-Santos A (2016) Anti-aging effects of guanosine in glial cells. Purinergic Signal 12(4):697–706
Nanetti L, Raffaelli F, Vignini A, Perozzi C, Silvestrini M, Bartolini M, Provinciali L, Mazzanti L (2011) Oxidative stress in ischaemic stroke. Eur J Clin Investig 41(12):1318–1322
Hansel G, Ramos DB, Delgado CA, Souza DG, Almeida RF, Portela LV, Quincozes-Santos A, Souza DO (2014) The potential therapeutic effect of guanosine after cortical focal ischemia in rats. PLoS One 9(2):e90693
Hansel G, Tonon AC, Guella FL, Pettenuzzo LF, Duarte T, Duarte MMMF, Oses JP, Achaval M, Souza DO (2015) Guanosine protects against cortical focal ischemia. Involvement of inflammatory response. Mol Neurobiol 52(3):1791–1803
de Oliveira DL et al (2004) Quinolinic acid promotes seizures and decreases glutamate uptake in young rats: reversal by orally administered guanosine. Brain Res 1018(1):48–54
Moretto MB, Arteni NS, Lavinsky D, Netto CA, Rocha JBT, Souza DO, Wofchuk S (2005) Hypoxic-ischemic insult decreases glutamate uptake by hippocampal slices from neonatal rats: prevention by guanosine. Exp Neurol 195(2):400–406
Molz S, Dal-Cim T, Budni J, Martín-de-Saavedra MD, Egea J, Romero A, del Barrio L, Rodrigues ALS, López MG, Tasca CI (2011) Neuroprotective effect of guanosine against glutamate-induced cell death in rat hippocampal slices is mediated by the phosphatidylinositol-3 kinase/Akt/glycogen synthase kinase 3beta pathway activation and inducible nitric oxide synthase inhibition. J Neurosci Res 89(9):1400–1408
Di Liberto V et al (2016) The guanine-based purinergic system: the tale of an orphan neuromodulation. Front Pharmacol 7:158
Volpini R, Marucci G, Buccioni M, Dal Ben D, Lambertucci C, Lammi C, Mishra RC, Thomas A, Cristalli G (2011) Evidence for the existence of a specific g protein-coupled receptor activated by guanosine. ChemMedChem 6(6):1074–1080
Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB (2000) Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol 57(4):667–678
Crossthwaite AJ, Hasan S, Williams RJ (2002) Hydrogen peroxide-mediated phosphorylation of ERK1/2, Akt/PKB and JNK in cortical neurones: dependence on Ca(2+) and PI3-kinase. J Neurochem 80(1):24–35
Brazil DP, Yang ZZ, Hemmings BA (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29(5):233–242
Niizuma K, Yoshioka H, Chen H, Kim GS, Jung JE, Katsu M, Okami N, Chan PH (2010) Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta 1802(1):92–99
Krizman-Genda E, González MI, Zelenaia O, Robinson MB (2005) Evidence that Akt mediates platelet-derived growth factor-dependent increases in activity and surface expression of the neuronal glutamate transporter, EAAC1. Neuropharmacology 49(6):872–882
Sims KD, Straff DJ, Robinson MB (2000) Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporter through activation of phosphatidylinositol 3-kinase. J Biol Chem 275(7):5228–5237
Frizzo ME, Frizzo JK, Amadio S, Rodrigues JM, Perry ML, Bernardi G, Volonté C (2007) Extracellular adenosine triphosphate induces glutamate transporter-1 expression in hippocampus. Hippocampus 17(4):305–315
Funding
Research was supported by grants from the Brazilian funding agencies: CAPES (Coordenação do Pessoal de Ensino Superior)—Project CAPES-PVE 052/2012; CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico)—Project INCT for Excitotoxicity and Neuroprotection; and FAPESC (Fundação de Amparo à Pesquisa e Inovação do Estado de Santa Catarina)—Project NENASC. C.I.T. is recipient of CNPq productivity fellowship.
Author information
Authors and Affiliations
Contributions
All authors have materially participated in the research and/or article preparation.
Corresponding author
Ethics declarations
The procedures used in the present study complied with the guidelines on animal care of the UFSC Ethics Committee on the Use of Animals (CEUA), which follows the Principles of laboratory animal care from NIH (2011).
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Dal-Cim, T., Poluceno, G.G., Lanznaster, D. et al. Guanosine prevents oxidative damage and glutamate uptake impairment induced by oxygen/glucose deprivation in cortical astrocyte cultures: involvement of A1 and A2A adenosine receptors and PI3K, MEK, and PKC pathways. Purinergic Signalling 15, 465–476 (2019). https://doi.org/10.1007/s11302-019-09679-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-019-09679-w