
Abstract
T cells respond to antigen stimulation with the rapid release of cellular ATP, which stimulates an autocrine feedback mechanism that regulates calcium influx through P2X receptors. This autocrine purinergic feedback mechanism plays an essential role in the activation of T cells resulting in cell proliferation and clonal expansion. We recently reported that increases in mitochondrial ATP production drive this stimulation-induced purinergic signaling mechanism but that low-level mitochondrial ATP production fuels basal T cell functions required to maintain vigilance of unstimulated T cells. Here we studied whether defects in these purinergic signaling mechanisms are involved in the unwanted proliferation of leukemia T cells. We found that acute leukemia T cells (Jurkat) possess a larger number and more active mitochondria than their healthy counterparts. Jurkat cells have higher intracellular ATP concentrations and generat more extracellular ATP than unstimulated T cells from healthy donors. As a result, increased purinergic signaling through P2X1 and P2X7 receptors elevates baseline levels of cytosolic Ca2+ in Jurkat cells. We found that pharmacological inhibition of this basal purinergic signaling mechanism decreases mitochondrial activity, Ca2+ signaling, and cell proliferation. Similar results were seen in the leukemic cell lines THP-1, U-937, and HL-60. Combined treatment with inhibitors of P2X1 or P2X7 receptors and the chemotherapeutic agent 6-mercaptopurine completely blocked Jurkat cell proliferation. Our results demonstrate that increased mitochondrial metabolism promotes autocrine purinergic signaling and uncontrolled proliferation of leukemia cells. These findings suggest that deranged purinergic signaling can result in T cell malignancy and that therapeutic targeting aimed at purinergic signaling is a potential strategy to combat T cell leukemia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute leukemia accounts for about 25,000 newly diagnosed cancer cases and 12,000 deaths annually in the USA alone [1]. About 25 % of these cases are classified as acute lymphoblastic leukemia (ALL). Progress in conventional chemotherapy of ALL has resulted in high cure rates of over 80 % in children. However, in adults, 5-year survival rates are between 45 and 50 % and prognosis is especially poor in cases with relapsing ALL [2–4]. Novel anti-cancer therapies are needed to improve outcome in adult ALL patients with chemotherapy-resistant disease.
Chemotherapeutic agents used in multi-drug regimens include methotrexate, 6-mercaptopurine, and fludarabine, all of which target purine metabolism [5]. The expression and function of enzymes in the purine pathway are often altered in ALL. In particular, T lymphoblasts overexpress adenosine deaminase (ADA) and have decreased purine nucleoside phosphorylase and 5′-ectonucleotidase (CD73) activity when compared to normal T cells [6]. Importantly, many of the nucleotide-metabolizing enzymes are also expressed on the cell surface of normal cells where they regulate extracellular concentrations of ATP and its hydrolysis products [7]. In this context, those ectoenzymes contribute to purinergic signaling processes that regulate the activation and function of T cells through autocrine feedback mechanisms that are fueled by cellular ATP release [8].
Normal T cells respond to the stimulation of T cell receptors (TCR) and CD28 co-receptors with the rapid release of cellular ATP that triggers purinergic receptors located on the cell surface of T cells [8–11]. Autocrine stimulation of P2X1, P2X4, and P2X7 receptors that function as ATP-gated ion channels triggers the influx of extracellular calcium ions (Ca2+), which is an essential step in the signaling process leading to IL-2 production and T cell effector functions [8, 11, 12]. Mitochondria generate the ATP that fuels this autocrine purinergic signaling process [13]. Here we show that human Jurkat leukemia cells differ from their normal CD4+ T cell counterparts in the purinergic signaling mechanisms that regulate cell activity. Leukemia cells possess a greater number and more active mitochondria than normal T cells, resulting in more active ATP release, higher pericellular ATP concentrations, and more robust autocrine purinergic stimulation of P2X1 and P2X7 receptors. We show that this unhinged purinergic signaling mechanism is responsible for continuous activation and unchecked proliferation of leukemia cells and that pharmacological inhibition of this dysregulated signaling cascade decreases mitochondrial activity and proliferation of leukemia cells. Our findings suggest that these purinergic signaling events are a potential pharmacological target for the treatment of T cell leukemia.
Materials and methods
Reagents
Dihydrorhodamine 123 (DHR123), carboxyfluorescein diacetate succinimidyl ester (CFSE), Fluo-4 AM, Fura-2 AM, SYTOX Green, tetramethylrhodamine ethyl ester (TMRE), and MitoTracker Green FM were purchased from Molecular Probes (Thermo Fisher Scientific, Waltham, MA). Dynabeads (Invitrogen, Thermo Fisher Scientific) coated with goat anti-mouse IgG antibodies were labeled with mouse anti-human CD3 and CD28 antibodies (BD Biosciences, San Jose, CA) and used for T cell stimulation. Suramin, NF023, NF157, NF340, 5-BDBD, A438079, 10panx1, and 6-mercaptopurine (6-MP) were purchased from Tocris Bioscience (Ellisville, MI). All other reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Cells
The human lymphoblastic T cell line Jurkat (clone E6-1; American Type Culture Collection, Manassas, VA), the acute monocytic leukemia cell line THP-1 (ATCC), and the histiocytic lymphoma cell line U-937 (ATCC) were maintained in RPMI-1640 medium (ATCC) supplemented with 10 % heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco, Thermo Fisher Scientific) at 37 °C in 5 % CO2. The acute promyelocytic leukemia cell line HL-60 (ATCC) was cultured in Iscove’s modified Dulbecco’s medium (ATCC) containing 20 % fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human CD4+ T cells were isolated from the blood of healthy subjects as previously described [14]. For some experiments, CD4+ T cells were stimulated for 4 h with anti-CD3/CD28-coated beads at a cell/bead ratio of 1:1. All experiments involving human subjects were approved by the Institutional Review Board of Beth Israel Deaconess Medical Center and informed written consent was obtained.
ATP hydrolysis, intracellular ATP content, and ATP release
To assess their ATP-hydrolyzing capability, Jurkat cells and purified CD4+ T cells (5 × 105) were incubated with 5 μM ATP for the indicated periods of time, cell-free culture supernatants were harvested on ice, and the concentrations of ATP and its hydrolysis products ADP, AMP, and adenosine were determined after etheno-derivatization of adenine compounds using high performance liquid chromatography (HPLC) as previously described [15]. For the assessment of intracellular ATP content, cells were snap-frozen in liquid nitrogen, sonicated in the presence of 0.4 M perchloric acid, and analyzed by HPLC. ATP release from Jurkat cells (5 × 105) treated for 10 min with carbonyl cyanide m-chlorophenylhydrazone (CCCP; 10 μM), rotenone (10 μM), carbenoxolone (CBX; 50 μM), or the pannexin-1 (panx1) mimetic inhibitory peptide 10panx1 (50 μM) was analyzed with a luciferin/luciferase ATP bioluminescence kit (Invitrogen).
Quantitative real-time PCR
Total RNA was extracted from Jurkat cells or CD4+ T cells (1 × 106) using the RNeasy Mini Kit (Qiagen, Valencia, CA) and converted to cDNA (QuantiTect reverse transcription kit, Qiagen) following the manufacturer’s protocol. Real-time PCR was performed on a Mastercycler® ep realplex instrument (Eppendorf, Hamburg, Germany) using iQTM SYBR® Green supermix (Bio-Rad, Hercules, CA) and QuantiTect primer assays (Qiagen). Expression levels of ectonucleoside triphosphate diphosphohydrylase (ENTPD) 1, ENTPD2, ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) 1, ENPP2, and ENPP3 were normalized to TATA-box binding protein (TBP), and the comparative Ct method was used for relative quantification of gene expression.
Imaging of mitochondrial membrane potential, reactive oxygen species (ROS) production, and pericellular ATP
Fluorescence live-cell imaging was performed with an inverted Leica DMI6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with a temperature-controlled (37 °C) stage incubator (Ibidi, Martinsried, Germany) and a Leica DFC365 FX camera. Cells were placed into fibronectin-coated (40 μg/ml) eight-well glass bottom chamber slides (Lab-Tek, Rochester, NY). Cells were treated or not treated with suramin (100 μM) or CCCP (10 μM) for 10 min, and mitochondrial membrane potential and ROS production were assessed by staining cells for 5 min with TMRE (100 nM) or DHR123 (10 μM), respectively. Fluorescence images were captured through a ×100 oil objective (NA 1.3, Leica) using TRITC and FITC filter sets (Leica Microsystems) and LeicaLAS microscope imaging software. ATP at the cell surface was imaged in Jurkat cells or CD4+ T cells suspended in HBSS and stained for 5 min with 500 nM of the cell surface targeting fluorescent ATP probe 2-2Zn(II) [16] using an YFP-2427A filter set (Semrock, Rochester, NY) and a Spot Boost EMCCD BT 2100 camera (Diagnostic Instruments Inc., Sterling Heights, MI). Images were analyzed with ImageJ software (National Institutes of Health).
Measurements of mitochondrial membrane potential, ROS production, and mitochondrial mass
Jurkat cells or primary CD4+ T cells were stained with DHR123 (10 μM), TMRE (100 nM), or MitoTracker Green (50 nM) for 15 min at 37 °C, and mitochondrial membrane potential, ROS production, and mitochondrial mass were measured using a FACScalibur flow cytometer (BD Biosciences, San Diego, CA) as previously described [17]. In some experiments, Jurkat cells were treated with inhibitors of P2X receptors at the indicated concentrations for 10 min prior to staining with DHR123 or TMRE.
Cytosolic Ca2+ measurements
Jurkat cells (5 × 105/ml) were treated for 10 min with suramin (200 μM) or vehicle control, stained with the cytosolic Ca2+ indicator Fluo-4 (5 μM) for 30 min at 37 °C, and analyzed by flow cytometry. To study the effect of P2X receptor blockade on basal Ca2+ levels in real time, Jurkat cells were loaded with Fura-2 AM (4 μM) for 30 min at 37 °C, washed twice, and resuspended in HBSS containing 0.1 % bovine serum albumin. Changes in cytosolic Ca2+ levels following addition of 200 μM suramin were monitored with a spectrofluorophotometer. The excitation wavelengths were 340 and 380 nm and emission was detected at 510 nm.
Resazurin/resorufin proliferation assay
For screening purposes, proliferation of Jurkat cells was assessed by measuring the reduction of resazurin to resorufin [18]. Cells (2.5 × 105/ml) were cultured in 96-well plates for 1 to 5 days as indicated. During the last 6 h of culture, cells were exposed to resazurin (100 μM), and conversion of resazurin to resorufin was assessed by measuring the absorbance at 570 nm (reduced state) and 600 nm (oxidized state) using a multimode plate reader (SpectraMax M5, Molecular Devices, Sunnyvale, CA). Specific absorbance was calculated by subtracting the absorbance at 600 nm from the absorbance at 570 nm, and the cell number in each well was estimated by comparing these values with a standard curve derived from wells with known cell numbers treated in parallel.
Flow cytometric analysis of proliferation
Cells (2.5 × 105/ml) were cultured in 96-well plates for 72 h, and the cell number in each well was determined using PE-labeled counting beads (BD Biosciences). To study cell division, Jurkat cells (5 × 106/ml) were labeled with CFSE (10 μM) and dilution of CFSE fluorescence was analyzed by flow cytometry. With each cell division, the fluorescently tagged proteins are evenly distributed among the daughter cells and the mean fluorescence halves.
Cell viability
Jurkat cells (2.5 × 105/ml) were seeded onto fibronectin-coated eight-well glass bottom chamber slides and cultured in the presence or absence of suramin (200 μM) or 6-MP (500 nM) at 37 °C and 5 % CO2. Cell condition was assessed after 72 h. Cells were labeled with propidium iodide (1.5 μM) to visualize dead cells, and bright field and fluorescence images (TRITC filter set) were captured with the microscope system described above using a ×40 objective (0.75/PH2, Leica). To quantify the percentage of dead cells, cells (2.5 × 105/ml) cultured in 96-well plates for 72 h in the presence or absence of the indicated drugs were stained with SYTOX Green (10 nM) for 15 min at 37 °C and analyzed by flow cytometry.
Statistics
Unless otherwise stated, data are expressed as mean values ± standard deviation (SD) of n ≥ 3 independent experiments. Statistical analyses were done using unpaired Student’s t test or non-parametric Mann–Whitney U test for two groups and one-way ANOVA followed by Holm-Sidak’s test or non-parametric Kruskal-Wallis test followed by Dunnett’s test for multiple comparisons. Differences were considered statistically significant at p < 0.05.
Results
Jurkat cells generate an ATP-rich extracellular environment
Solid tumors are thought to generate a tumor microenvironment that promotes cell proliferation and immune evasion [19]. High concentrations of ATP and its breakdown products in the tumor interstitium may favor cancer growth [20, 21]. Because extracellular ATP contributes to the proliferation of normal T cells, we wondered whether elevated extracellular ATP concentrations drive the continuous activation and proliferation of leukemia T cells. In order to test this possibility, we compared the concentrations of ATP and its breakdown products in the culture supernatants of Jurkat cells and of primary CD4+ T cells from healthy human subjects. We found that Jurkat cell cultures generate about three times more ATP and ADP than their healthy counterparts (Fig. 1a). Stimulation of primary T cells with anti-CD3/anti-CD28 antibody-coated beads increased the concentration of extracellular ATP, which is consistent with the notion that ATP promotes T cell activation. Next, we studied whether altered expression levels of ectonucleotidases that are responsible for the enzymatic breakdown of extracellular ATP may be the reason for these differences between normal and leukemic T cells. Except for ENPP-1, the mRNA expression levels of all ectonucleotidases that convert ATP to ADP, AMP, or adenosine were significantly lower in Jurkat cells than in primary CD4+ T cells (Fig. 1b). Interestingly, a similar trend towards decreased ectonucleotidase expression levels was seen in healthy CD4+ T cells in response to cell stimulation, suggesting that downregulation of ATP-hydrolyzing ectoenzymes and decreased ATP breakdown contribute to the activation and proliferation of T cells (Fig. 1b). To test this notion, we compared the capacity of Jurkat and healthy T cells to hydrolyze extracellular ATP. Equal numbers of Jurkat and healthy cells were incubated with ATP (5 μM) and the kinetics of ATP breakdown by these cells were compared (Fig. 1c–f). Consistent with the higher expression of ectonucleotidases in healthy CD4+ T cells, healthy cells were more efficient than Jurkat cells in eliminating extracellular ATP (Fig. 1c). Jurkat cells converted ATP mostly to ADP (Fig. 1d), while healthy CD4+ T cells formed primarily AMP (Fig. 1e) and adenosine (Fig. 1f). Taken together, these results demonstrate that diminished expression of ectonucleotidases in Jurkat cells reduces their ability to decrease extracellular ATP concentrations, which may result in an ATP-rich extracellular environment and promote leukemia cell growth.

Ectonucleotidase expression contributes to an ATP-rich extracellular environment in Jurkat cells. a Concentrations of ATP, ADP, AMP, and adenosine (ADO) in the cell culture supernatants of Jurkat cells (n = 9), healthy primary CD4+ T cells (n = 11), or CD4+ T cells stimulated for 5 min with anti-CD3/anti-CD28-coated beads (n = 3) were analyzed by HPLC; ***p < 0.001 (Student’s t test). b mRNA expression pattern of ectonucleotidases in Jurkat cells or normal CD4+ T cells stimulated or not for 4 h with anti-CD3/CD28-coated beads. The primary degradation product is indicated by arrows. Results are expressed as mean ± SD of n = 3–4 independent experiments. *p < 0.05 (one-way ANOVA); n.d., not detected; ENTPD, ectonucleoside triphosphate diphosphohydrylase; ENPP, ectonucleotide pyrophosphatase/phosphodiesterase; TBP, TATA-box binding protein. c–f 5 μM ATP was added to Jurkat or primary CD4+ T cell cultures, and the concentrations of ATP (c), ADP (d), AMP (e), and adenosine (f) in the supernatants were determined by HPLC. Data show mean values ± SD of n = 2–3 separate experiments
Leukemic T cells generate more mitochondrial ATP than healthy T cells
Normal T cells respond to TCR/CD28 stimulation with the rapid release of cellular ATP into the extracellular environment [11–13]. However, unstimulated T cells also release ATP, albeit at a lower rate than stimulated cells [22]. We found that this continuous basal ATP release feeds a basal purinergic feedback loop that maintains T cell vigilance and the ability of T cells to rapidly respond to external stimuli [17]. Therefore, we wondered whether excessive activity of this basal purinergic feedback mechanism is associated with the unchecked proliferation of leukemia cells. Using a novel membrane-anchoring fluorescence probe, termed 2-2Zn(II) and developed in the laboratory of Dr. Hamachi, Kyoto, Japan [16], we found that the extracellular ATP concentration at the cell surface of Jurkat cells was considerably higher than that of healthy CD4+ T cells (Fig. 2a). These findings confirm that pericellular ATP concentrations are higher in leukemia cells than in their healthy counterparts, either due to increased release of ATP, reduced hydrolysis by ectonucleotidases, or both.

Jurkat cells produce and release more mitochondrial ATP than normal T cells. a Pericellular ATP in Jurkat cells or primary CD4+ T cells was visualized with the membrane-bound fluorescent ATP probe 2-2Zn(II); ×100 oil objective; scale bar, 5 μm. b Jurkat cells were treated with CBX (50 μM), the pannexin-1 specific inhibitory peptide 10panx1 (50 μM), CCCP (10 μM), or rotenone (10 μM) for 10 min and extracellular ATP (eATP) concentrations in the bulk supernatant were measured with a bioluminescence kit. Data represent means ± SD of n = 3 separate experiments; *p < 0.05 (one-way ANOVA). c Intracellular concentrations of ATP, ADP, AMP, and adenosine (ADO) of Jurkat (n = 9) and healthy CD4+ T cells (n = 8) were determined by HPLC. Results are expressed as mean ± SD; ***p < 0.001, *p < 0.05 (Student’s t test). d Mitochondrial content was assessed using MitoTracker Green staining and flow cytometry. A representative histogram is shown in the panel on the left and cumulative results of n = 5–6 independent experiments are shown on the right; **p < 0.01 (Mann–Whitney U test). e, f Cells were stained with TMRE to assess mitochondrial membrane potential (ΔΨm) and with DHR123 to assess mitochondrial reactive oxygen species (ROS) formation using fluorescence microscopy (e; ×100 oil objective, NA 1.3; scale bar, 5 μm) and flow cytometry (f). Data represent mean values ± SD of n = 6 (TMRE) or n = 4 (DHR123) independent experiments; **p < 0.01, *p < 0.05 (Mann–Whitney U test); MFI, mean fluorescence intensity
Mitochondria generate the majority of the ATP that normal T cells release into the extracellular space [13]. Therefore, we wondered whether mitochondria contribute to the robust ATP release from Jurkat cells. We treated Jurkat cells for 10 min with rotenone, an inhibitor of complex I of the electron transport chain or with CCCP, an uncoupler of oxidative phosphorylation. Rotenone and CCCP significantly reduced extracellular ATP concentrations in Jurkat cell cultures (Fig. 2b). Inhibition of panx1 channels that facilitate cellular ATP release with the inhibitors CBX or 10panx1 had a similar effect on ATP release from Jurkat cells (Fig. 2b). We also found that intracellular ATP concentrations in normal CD4+ T cells were significantly lower than in Jurkat cells (Fig. 2c). Cancer cells are thought to preferentially utilize glycolytic pathways rather than oxidative respiration for energy production [23]. In order to compare the abundance of mitochondria in Jurkat cells and healthy CD4+ T cells, we estimated mitochondrial mass with MitoTracker Green. MitoTracker Green staining is widely used and accepted for this purpose [24]; however, some concerns were raised regarding a possible influence of mitochondrial membrane potential (ΔΨm) on this dye [25]. To test this possibility, we stained cells with MitoTracker Green and disrupted ΔΨm with CCCP. MitoTracker Green staining was largely independent of ΔΨm under our experimental conditions (Supplementary Fig. 1). Using MitoTracker Green staining, we found that Jurkat cells possess ~3 times more mitochondrial mass than their healthy counterparts and that mitochondria in Jurkat cells are significantly more active compared to unstimulated CD4+ T cells of healthy subjects (Fig. 2d). The latter conclusion was supported by a higher mitochondrial membrane potential (ΔΨm) and increased mitochondrial ROS production in Jurkat cells when compared to CD4+ T cells (Fig. 2e, f). Taken together, these results suggest that a larger number of active mitochondria produce extracellular ATP concentrations that promote excessive activation and proliferation of leukemia cells.
Autocrine purinergic signaling perpetuates mitochondrial activity in Jurkat cells
The activation of normal CD4+ T cells triggers ATP release, P2 receptor stimulation, Ca2+ influx, and mitochondrial firing to set in motion a signaling loop that culminates in T cell proliferation [11–13]. Excessive ATP release from cancer cells may propagate this signaling loop and thereby contribute to the perpetual proliferation of neoplastic T cells. In support of this notion, we found that the P2 receptor antagonist suramin reduced cytosolic Ca2+ levels, ΔΨm, and mitochondrial ROS production of Jurkat cells (Fig. 3a–d). The effects of suramin were paralleled by similar effects of CCCP (Fig. 3c, d), suggesting that mitochondrial ATP production, ATP release, and P2 receptor stimulation maintain upregulated basal mitochondrial activity levels that promote proliferation of cancer cells.

Autocrine purinergic signaling maintains mitochondrial activity in Jurkat cells. a Basal cytosolic Ca2+ levels in Jurkat cells were measured by flow cytometry after labeling cells with Fluo-4 AM in the presence or absence of suramin (200 μM). The panel on the left shows a representative histogram and cumulative results of n = 3–4 experiments are shown in the panel on the right. **p < 0.01 (Student’s t test); MFI, mean fluorescence intensity. b Jurkat cells were loaded with Fura-2 AM and cytosolic Ca2+ was monitored with a spectrofluorometer for 90 s. Suramin (200 μM) or vehicle control was added at 45 s (arrow). Data are expressed as the ratio of fluorescence signals detected at 510 nm with excitation at 340 and 380 nm and are representative of n = 4 experiments with similar results. c, d Cells were treated with suramin (200 μM), CCCP (10 μM), or vehicle control for 10 min. Then they were stained for another 5 min with TMRE to assess the mitochondrial membrane potential (ΔΨm; c) or with DHR123 to assess mitochondrial reactive oxygen species (ROS) formation (d) and analyzed by fluorescence microscopy. Results in the panels on the right are expressed as mean gray values ± SD of n > 20 cells of n ≥ 3 experiments; *p < 0.05; Kruskal-Wallis test (c) or one-way ANOVA (d). ×100 objective, NA 1.3, scale bar, 10 μm
P2X1 and P2X7 receptors maintain mitochondrial activity in Jurkat cells
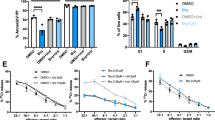
Next we asked which P2 receptors are involved in the feedback loop that maintains these elevated levels of basal mitochondrial activity in Jurkat cells. Earlier studies have shown that P2X1, P2X4, and P2X7 receptors initiate cell activation in response to T cell stimulation via TCR/CD28 receptors [11, 12]. Using antagonists of these receptors (NF157 or NF023 for P2X1; 5-BDBD for P2X4; A438079 for P2X7; and suramin for all P2 receptors), we found that P2X1 and to a lesser degree P2X7 receptors maintain basal mitochondrial activity in unstimulated Jurkat cells (Fig. 4a, b). The P2X1 antagonists NF157 and NF023 were nearly as potent as suramin in inhibiting mitochondrial ROS production (Fig. 4a–c). Because NF157 inhibits P2X1 but also P2Y11 receptors, we also tested the more specific P2Y11 receptor antagonist NF340 and found that this inhibitor had no effect on mitochondrial ROS production (Fig. 4c). Taken together, these results suggest that P2X1 and P2X7 receptors are central to maintaining the vigorous basal purinergic feedback loop that is responsible for perpetual mitochondrial activation in Jurkat cells.

P2X1 and P2X7 receptors maintain mitochondrial activity in Jurkat cells. a Jurkat cells were treated for 10 min with the indicated concentrations of the general P2 receptor inhibitor suramin, stained with TMRE or DHR123 and mitochondrial membrane potential (ΔΨm) and reactive oxygen species (ROS) production were analyzed by flow cytometry. b, c Jurkat cells were treated for 10 min with the indicated concentrations of the specific P2 receptor antagonists NF023 (P2X1), 5-BDBD (P2X4), A438079 (P2X7), NF157 (P2X1 and P2Y11), or NF340 (P2Y11), stained with DHR123, and mitochondrial ROS formation was assessed by flow cytometry. Data show mean values ± SD of n = 3–6 experiments. Results in b and c are expressed as percentage of cells with active mitochondria where the mitochondrial ROS production of untreated cells was set to 100 % and the ROS production of suramin (100 μM)-treated cells to 0 %; *p < 0.05 vs. non-inhibitor treated control samples (one-way ANOVA); MFI, mean fluorescence intensity
Perpetual stimulation of P2X1 and P2X7 receptors maintains the growth of leukemia cells
Next, we tested the concept that perpetual stimulation of P2 receptors drives the proliferation of cancer cells. Stimulation with the non-hydrolysable ATP analogue ATPγS (100 μM) increased the proliferation of Jurkat cells only slightly beyond the level seen in untreated Jurkat cells (Fig. 5a). However, removal of extracellular ATP with apyrase significantly diminished cell proliferation. Similarly, blocking ATP release with the gap junction inhibitor CBX or the panx1-specific blocker 10panx1 significantly inhibited the proliferation of Jurkat cells. These findings support the notion that continuous basal ATP release and autocrine purinergic signaling maintain the unbridled proliferation of leukemia cells. In agreement with this concept, we found that inhibition of P2 receptor signaling with suramin or of mitochondrial ATP formation with CCCP profoundly suppressed Jurkat cell proliferation (Fig. 5a). The anti-proliferative effects of CBX and suramin resulted in significantly lower cell numbers 48 h after inhibition of basal purinergic signaling in Jurkat cells (Fig. 5b). CBX and suramin dose-dependently reduced Jurkat cell proliferation (Fig. 5c). Treatment of Jurkat cells with the P2X1 receptor antagonists NF023 and NF157 or the P2X7 receptor antagonist A438079 recapitulated the anti-proliferative effect of suramin (Fig. 5d), supporting the concept that continuous stimulation of P2X1 and P2X7 receptors by basal ATP release perpetuates unimpeded proliferation of leukemia cells.

P2X1 and P2X7 receptor signaling perpetuates Jurkat cells proliferation. a Jurkat cells were cultured in the presence or absence of ATPγS (100 μM), apyrase (40 U/ml), CBX (50 μM), 10panx1 (50 μM), suramin (200 μM), or CCCP (1 μM) for 72 h and proliferation was assessed by measuring the reduction of resazurin to resorufin. b Jurkat cells were cultured for the indicated periods of time with suramin (200 μM), CBX (100 μM), or vehicle control, and reduction of resazurin was used to calculate cell numbers in each well. c, d Jurkat cells were cultured in the presence or absence of the indicated concentrations of suramin or CBX (c) or the specific P2X inhibitors NF023 (P2X1), NF157 (P2X1, P2Y11), or A438079 (P2X7) for 72 h and cells were counted with a flow cytometer. Data show mean values ± SD of n = 4 experiments. *p < 0.05 vs. non-inhibitor treated controls (one-way ANOVA)
Proliferation of various other leukemia cell types also involves P2 receptor signaling
In order to test whether basal ATP release sustains the proliferation of other leukemia cell types, we studied the effect of CBX and suramin on THP-1 (acute monocytic leukemia), U-937 (histiocytic lymphoma), and HL-60 (acute promyelocytic leukemia) cells. Suramin inhibited proliferation of all three cell types to varying degrees (Fig. 6a–c). The effect of suramin on these cells was however weaker when compared to Jurkat cells (Fig. 6c). Similarly, the sensitivity of cells to CBX differed between the four leukemia cell types. Jurkat and U-937 cells were most sensitive to CBX treatment (Fig. 6b), while proliferation of HL-60 and THP-1 cells was only moderately inhibited by CBX (Fig. 6a, c ). While the extracellular ATP levels were also different among the four cell lines, they were three to five times higher than the ATP levels in primary healthy CD4+ T cell cultures (Fig. 6d). In summary, these results suggest that cell growth of these different leukemia cell lines involves basal ATP release and autocrine purinergic signaling, albeit to varying degrees.

Leukemic cell growth depends to varying degrees on ATP release and P2 receptor signaling. THP-1 (a), U-937 (b), and HL-60 (c) cells were cultured in the presence or absence of the indicated concentrations of CBX or suramin for 72 h and cells were counted with a flow cytometer. Data are expressed as mean ± SD of n = 4 experiments. *p < 0.05 vs. non-inhibitor treated controls (one-way ANOVA). d Cells as indicated (5 × 105/200 μl) were incubated for 30 min at 37 °C on a vibration isolation table and extracellular ATP (eATP) concentrations in the supernatant were measured with a bioluminescence kit. Data represent means ± SD of n = 3 experiments; *p < 0.05 (one-way ANOVA)
Inhibition of purinergic signaling as anti-proliferative adjuvant treatment strategy
The anti-cancer drug 6-mercaptopurine (6-MP) is commonly used in multi-drug regimens for the treatment of ALL. We found that 6-MP (500 nM) and suramin (200 μM) had similar anti-proliferative effects on Jurkat cells, reducing cell growth by up to 80 % compared to untreated controls (Fig. 7a, b). Importantly, the combination of suramin and 6-MP increased the anti-proliferative potential of each single drug and completely prevented cell proliferation, resulting in a net loss of cells after 72 h in culture (Fig. 7b). Comparison of 6-MP and suramin revealed similar cytostatic and cytotoxic properties that were amplified by combined treatment of Jurkat cells with both drugs (Fig. 7c, d). Combinations of 6-MP with specific inhibitors of P2X1 (NF023) or P2X7 (A438079) receptors evoked similar but slightly weaker responses than the combination of 6-MP with suramin (Fig. 7b, d). These findings suggest that both P2X1 and P2X7 receptors are involved in the purinergic feedback mechanisms that maintain cell proliferation and that purinergic signaling mechanisms may be potential drug targets to increase the effectiveness of current chemotherapeutic regimens for ALL.

Inhibition of purinergic signaling amplifies the anti-proliferative activity of 6-mercaptopurine. a–d Jurkat cells were cultured for 72 h in the presence or absence of suramin (200 μM), 6-mercaptopurine (6-MP; 500 nM), NF023 (10 μM), A438079 (μM), or combinations of 6-MP and the various P2 receptor inhibitors. a Cells were stained with propidium iodide (PI) and cell density and PI-positive (dead; red) cells were assessed by bright field and fluorescence microscopy. Images were merged and are representative for n > 15 microscopic fields from three separate experiments; ×40 objective, scale bar, 20 μm. b Cells were counted with a flow cytometer. Data represent means ± SD, n = 4–8; *p < 0.05 (one-way ANOVA). c Cell division was analyzed by measuring CFSE dilution. The dotted line indicates the main peak of control cells cultured for 72 h without drugs. The histograms shown are representative for n = 8 experiments with similar results. d The percentage of dead cells was determined using SYTOX Green staining and flow cytometry. Data represent means ± SD, n = 4; *p < 0.05 (one-way ANOVA)
Discussion
Cellular ATP release from neurons and non-excitable cells has become recognized as an important mechanism in the regulation of mammalian cell functions [26]. Normal T cells respond to TCR/CD28 stimulation with the rapid release of ATP that fuels autocrine signaling via P2X receptors to initiate effector functions including cell proliferation [8, 10–12]. Mitochondria are the main source of the ATP that drives this autocrine purinergic feedback process [13]. However, unstimulated T cells also rely on purinergic signaling. They are not inactive but maintain a level of vigilance that allows them to detect and respond to antigens in their environment. We have recently shown that unstimulated T cells maintain vigilance via a basal purinergic feedback mechanism that involves low-level mitochondrial activity and ATP production to activate P2X receptors and maintain cellular Ca2+ homeostasis [17]. In the present study, we found that this basal purinergic signaling mechanism is disturbed in Jurkat cells, resulting in the release of large amounts of ATP that promote proliferation without the need for TCR/CD28 stimulation.
Purinergic signaling mechanisms are a potential novel drug target for cancer therapy [27–29]. However, existing reports about the role of extracellular ATP in cancer cell growth are contradictory. Both anti-proliferative and growth-promoting effects have been ascribed to extracellular ATP; these differences may be due to differences in the expression profiles of the various P2 receptor subtypes that can have opposing effects on cell growth [27]. P2X7 receptor expression has been linked to cell proliferation [30]. Overexpression of P2X7 receptors increases cell proliferation of K562 and LG14 cells, two leukemic cell lines that lack endogenous P2X7 receptors [31]. Furthermore, P2X7 receptor expression in bone marrow-derived mononuclear cells is significantly higher in patients with ALL and other forms of leukemia than in healthy controls [32]. In B cell chronic lymphocytic leukemia, P2X7 receptor expression was shown to correlate with increased morbidity, which suggests that P2X7 receptors confer a growth advantage to neoplastic cells [33]. In agreement with those reports, we found that P2X7 receptors contribute to the proliferation of Jurkat T cells. However, our results also suggest that P2X1 receptors are more important than P2X7 receptors for Jurkat cell proliferation. Further studies using genetic approaches such as receptor silencing experiments will be needed to verify these results and to specify the respective contributions of individual P2X receptor subtypes to cell proliferation.
One of the key findings of our report is that mitochondrial activity is significantly higher in Jurkat cells than in their healthy counterparts, resulting in elevated extracellular ATP levels that stimulate P2X receptors and promote cancer cell proliferation. This finding demands reevaluation of the current view on mitochondria in cancer cell biology. Cancer cells are generally thought to make little use of mitochondria and to primarily rely on aerobic glycolysis for their energy demand [23, 34]. Our findings do not contradict this notion. However, while mitochondria are not essential for energy production in cancer cells, our findings show that mitochondria are a driving force that perpetuates proliferation and unimpeded growth of cancer cells. Recently, other evidence has emerged in support of a similar role for mitochondria in tumorigenesis [35–37]. We previously reported that mitochondria are central components of the purinergic signaling mechanisms that regulate immune cell functions [13, 17, 38, 39]. Our current results show that highly active mitochondria in Jurkat cells up-regulate the purinergic signaling processes that regulate cell metabolism and promote the proliferation of Jurkat cells.
We found that inhibition of purinergic signaling, for example with P2 receptor antagonists, disrupts mitochondrial metabolism and thus cuts off the cycle that maintains unimpeded cell proliferation. Treating cells with suramin significantly suppressed the growth of Jurkat cells. We saw a moderate increase in the number of dead cells; however, the exact underlying mechanisms by which suramin suppresses cell growth are not clear. Further studies are necessary to define the primary mode of action of suramin, which may include cell death via apoptosis, necroptosis, or mitochondrial permeability transition (MPT)-regulated processes [40]. Suramin also enhanced the anti-proliferative activity of low-dose 6-MP, a chemotherapeutic agent commonly used in ALL. The aim of combined chemotherapy is to maximize the anti-neoplastic activity while at the same time minimizing drug-associated side effects and the risk of drug resistance. In combination with suramin, 6-MP completely inhibited cell growth at a concentration that minimizes its side effects. The general P2 receptor antagonist suramin has been used for more than 80 years in the treatment of African trypanosomiasis (sleeping sickness) and onchocerciasis, also known as river blindness [41]. Over the last two decades, suramin has been studied for the treatment of solid tumors, including prostate, ovarian, and lung cancer [42–44]. While the anti-tumor activity of suramin was often ascribed to its ability to inhibit the binding of growth factors to their receptors [45, 46], additional mechanisms were suspected and are likely to involve purinergic signaling. In support of this notion, we found that the effect of suramin could be partially replicated by inhibitors of P2X1 and P2X7 receptors. Suramin was tested in clinical trials for prostate cancer, but these trials were stopped because of significant toxic side effects [47–49]. Our findings suggest that lower, less toxic doses of suramin or of specific P2X receptor inhibitors could be used in combination with other chemotherapeutic agents such as 6-MP.
Several lines of evidence suggest that purinergic signaling is a possible target for cancer therapy [20, 50, 51]. In fact, the adenosine deaminase inhibitor pentostatin is being used to treat specific forms of leukemia demonstrating the therapeutic potential of targeting purinergic signaling in leukemia [52, 53]. In support of a more general role for the identified growth-promoting purinergic signaling mechanisms, we found that monocytic or promyelocytic leukemia cell lines also depend on autocrine purinergic signaling for cell proliferation, albeit to different extents. The varying sensitivities to purinergic inhibitors of the different cancer cell lines could be explained by differences in mitochondrial function that were previously described in these different cancer cell lines [54]. However, the complex purinergic signaling events that regulate T cells are still not fully defined. Our current findings indicate that disturbances of the purinergic control mechanisms of T cell vigilance cause neoplastic behavior. However, different types of leukemia cells likely differ with regard to their individual defects. In this respect, the use of cell lines to model clinical disease is a clear limitation of our current study. Future studies will be necessary to determine the roles of basal ATP release and autocrine purinergic signaling in the growth and malignancy of primary leukemic CD4+ T cells from actual cancer patients. Such studies may help define a common denominator that is suitable for targeted treatment of a wider range of leukemia cells.
References
Siegel R, Ma J, Zou Z, Jemal A (2008) Cancer statistics, 2014. CA Cancer J Clin 64:9–29
Pui CH, Robison LL, Look AT (2008) Acute lymphoblastic leukaemia. Lancet 371:1030–1043
Pui CH (2009) T cell acute lymphoblastic leukemia: NOTCHing the way toward a better treatment outcome. Cancer Cell 15:85–87
Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM (2012) Acute leukemia incidence and patient survival among children and adults in the United States, 2001–2007. Blood 119:34–43
Riscoe MK, Brouns MC, Fitchen JH (1989) Purine metabolism as a target for leukemia chemotherapy. Blood Rev 3:162–173
Poplack DG, Blatt J, Reaman G (1981) Purine pathway enzyme abnormalities in acute lymphoblastic leukemia. Cancer Res 41:4821–4823
Zimmermann H, Zebisch M, Sträter N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8:437–502
Junger WG (2011) Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11:201–212
Filippini A, Taffs RE, Sitkovsky MV (1990) Extracellular ATP in T-lymphocyte activation: possible role in effector functions. Proc Natl Acad Sci U S A 87:8267–8271
Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, Verderio C, Buer J, Scanziani E, Grassi F (2008) Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal 1(39):ra6
Yip L, Woehrle T, Corriden R, Hirsh M, Chen Y, Inoue Y, Ferrari V, Insel PA, Junger WG (2009) Autocrine regulation of T-cell activation by ATP release and P2X7 receptors. FASEB J 23:1685–1693
Woehrle T, Yip L, Elkhal A, Sumi Y, Chen Y, Yao Y, Insel PA, Junger WG (2010) Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 116:3475–3484
Ledderose C, Bao Y, Lidicky M, Zipperle J, Li L, Strasser K, Shapiro NI, Junger WG (2014) Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling. J Biol Chem 289:25936–25945
Yip L, Cheung CW, Corriden R, Chen Y, Insel PA, Junger WG (2007) Hypertonic stress regulates T-cell function by the opposing actions of extracellular adenosine triphosphate and adenosine. Shock 27:242–250
Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, Inoue Y, Tanaka H, Junger WG (2014) Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock 42:142–147
Kurishita Y, Kohira T, Ojida A, Hamachi I (2012) Organelle-localizable fluorescent chemosensors for site-specific multicolor imaging of nucleoside polyphosphate dynamics in living cells. J Am Chem Soc 134:18779–18789
Ledderose C, Bao Y, Ledderose S, Woehrle T, Heinisch M, Yip L, Zhang J, Robson SC, Shapiro NI, Junger WG (2016) Mitochondrial dysfunction depletes purinergic signaling mechanisms needed for T cell vigilance and immune defense. J Infect Dis 213:456–464
Zhi-Jun Y, Sriranganathan N, Vaught T, Arastu SK, Ahmed SA (1997) A dye-based lymphocyte proliferation assay that permits multiple immunological analyses: mRNA, cytogenetic, apoptosis, and immunophenotyping studies. J Immunol Methods 210:25–39
Albini A, Sporn MB (2007) The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer 7:139–147
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M (2006) A2A adenosine receptor protects tumors from anti-tumor T cells. Proc Natl Acad Sci U S A 103:13132–13137
Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F (2008) Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS ONE 3(7), e2599
Yegutkin GG, Mikhailov A, Samburski SS, Jalkanen S (2006) The detection of micromolar pericellular ATP pool on lymphocyte surface by using lymphoid ecto-adenylate kinase as intrinsic ATP sensor. Mol Biol Cell 17:3378–3385
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Pendergrass W, Wolf N, Poot M (2004) Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 61:162–169
Keij JF, Bell-Prince C, Steinkamp JA (2000) Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry 39:203–210
Burnstock G, Fredholm BB, North RA, Verkhratsky A (2010) The birth and postnatal development of purinergic signaling. Acta Physiol 199:93–147
White N, Burnstock G (2006) P2 receptors and cancer. Trends Pharmacol Sci 27:211–217
Burnstock G, Di Virgilio F (2013) Purinergic signaling and cancer. Purinergic Signal 9:491–540
Antonioli L, Blandizzi C, Pacher P, Hasko G (2013) Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev 13:842–857
Di Virgilio F, Ferrari D, Adinolfi E (2009) P2X7: a growth-promoting receptor—implications for cancer. Purinergic Signal 5:251–256
Baricordi OR, Melchiorri L, Adinolfi E, Falzoni S, Chiozzi P, Buell G, Di Virgilio F (1999) Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J Biol Chem 274:33206–33208
Zhang XJ, Zheng GG, Ma XT, Yang YH, Li G, Rao Q, Nie K, Wu KF (2004) Expression of P2X7 in human hematopoietic cell lines and leukemia patients. Leuk Res 28:1313–1322
Adinolfi E, Melchiorri L, Falzoni S, Chiozzi P, Morelli A, Tieghi A, Cuneo A, Castoldi G, Di Virgilio F, Baricordi OR (2002) P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood 99:706–708
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Weinberg SE, Chandel NS (2015) Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 11:9–15
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698
Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313:459–465
Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, Junger WG (2014) Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem 289:26794–26803
Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, Junger WG (2015) mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol 210:1153–1164
Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M et al (2015) Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22:58–73
Voogd TE, Vansterkenburg ELM, Wilting J, Janssen LHM (1993) Recent research on the biological activity of suramin. Pharmacol Rev 45:177–203
La Rocca RV, Danesi R, Cooper MR, Jamis-Dow CA, Ewing MW, Linehan WM, Myers CE (1991) Effect of suramin on human prostate cancer cells in vitro. J Urol 145:393–398
Taylor CW, Lui R, Fanta P, Salmon SE (1992) Effects of suramin on in vitro growth of fresh human tumors. J Natl Cancer Inst 84:489–494
Rubio GJ, Pinedo HM, Virizuela J, van Ark-Otte J, Giaccone G (1995) Effects of suramin on human lung cancer cell lines. Eur J Cancer 31A:244–251
Hosang M (1985) Suramin binds to platelet-derived growth factor and inhibits its biological activity. J Cell Biochem 29:265–273
Pollak M, Richard M (1990) Suramin blockade of insulin-like growth factor I-stimulated proliferation of human osteosarcoma cells. J Natl Cancer Inst 82:1349–1352
Figg WD, Cooper MR, Thibault A, Headlee D, Humphrey J, Bergan RC, Reed E, Sartor O (1994) Acute renal toxicity associated with suramin in the treatment of prostate cancer. Cancer 74:1612–1614
Dawson NA, Cooper MR, Figg WD, Headlee DJ, Thibault A, Bergan RC, Steinberg SM, Sausville EA, Myers CE, Sartor O (1995) Antitumor activity of suramin in hormone-refractory prostate cancer controlling for hydrocortisone treatment and flutamide withdrawal as potentially confounding variables. Cancer 76:452–463
Small EJ, Meyer M, Marshall ME, Reyno LM, Meyers FJ, Natale RB, Lenehan PF, Chen L, Slichenmyer WJ, Eisenberger M (2000) Suramin therapy for patients with symptomatic hormone-refractory prostate cancer: results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydrocortisone. J Clin Oncol 18:1440–1450
Beavis PA, Stagg J, Darcy PK, Smyth MJ (2012) CD73: a potent suppressor of antitumor immune response. Trends Immunol 33:231–237
Antonioli L, Blandizzi C, Pacher P, Haskó G (2013) Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 13:842–857
Robak P, Robak T (2013) Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat Rev 39:851–861
Salam L, Abdel-Wahab O (2015) Hairy cell leukemia: update and current therapeutic approach. Curr Opin Hematol 22:355–361
Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatin DM (2014) Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508:108–112
Acknowledgments
This work was funded in part by grants from the National Institutes of Health, GM-51477, GM-60475, AI-080582, and T32GM103702 (W.G.J.), and from the German Research Foundation (DFG), LE-3209/1-1 (C.L.). We thank Drs. Yasutaka Kurishita and Itaru Hamachi for kindly providing the fluorescent ATP probe 2-2Zn(II).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Fig. 1
Staining of mitochondria in Jurkat cells with MitoTracker Green does not depend on mitochondrial membrane potential. a–c Jurkat cells were treated with CCCP (1 μM) or HBSS (control) for 10 min, loaded with MitoTracker Green AM (50 nM) or TMRE (100 nM) for 15 min and analyzed with a flow cytometer. Representative histograms are shown in panels a and b and cumulative results of n = 3 experiments (mean ± SD) are shown in panel c. MTG, MitoTracker Green; MFI, mean fluorescence; n.s., non-significant; ***p < 0.001, t test. (GIF 35 kb)
Rights and permissions
About this article

Cite this article
Ledderose, C., Woehrle, T., Ledderose, S. et al. Cutting off the power: inhibition of leukemia cell growth by pausing basal ATP release and P2X receptor signaling?. Purinergic Signalling 12, 439–451 (2016). https://doi.org/10.1007/s11302-016-9510-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-016-9510-y