
Abstract
Glioblastoma multiforme (GBM), the most common and aggressive brain tumor in humans, comprises a population of stem-like cells (GSCs) that are currently investigated as potential target for GBM therapy. Here, we used GSCs isolated from three different GBM surgical specimens to examine the antitumor activity of purines. Cultured GSCs expressed either metabotropic adenosine P1 and ATP P2Y receptors or ionotropic P2X7 receptors. GSC exposure for 48 h to 10–150 μM ATP, P2R ligand, or to ADPβS or MRS2365, P2Y1R agonists, enhanced cell expansion. This effect was counteracted by the PY1R antagonist MRS2500. In contrast, 48-h treatment with higher doses of ATP or UTP, which binds to P2Y2/4R, or 2′(3′)-O-(4-benzoylbenzoyl)-ATP (Bz-ATP), P2X7R agonist, decreased GSC proliferation. Such a reduction was due to apoptotic or necrotic cell death but mostly to growth arrest. Accordingly, cell regrowth and secondary neurosphere formation were observed 2 weeks after the end of treatment. Suramin, nonselective P2R antagonist, MRS1220 or AZ11645373, selective A3R or P2X7R antagonists, respectively, counteracted ATP antiproliferative effects. AZ11645373 also abolished the inhibitory effect of Bz-ATP low doses on GSC growth. These findings provide important clues on the anticancer potential of ligands for A3R, P2Y1R, and P2X7R, which are involved in the GSC growth control. Interestingly, ATP and BzATP potentiated the cytotoxicity of temozolomide (TMZ), currently used for GBM therapy, enabling it to cause a greater and long-lasting inhibitory effect on GSC duplication when readded to cells previously treated with purine nucleotides plus TMZ. These are the first findings identifying purine nucleotides as able to enhance TMZ antitumor efficacy and might have an immediate translational impact.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Grade IV astrocytoma or glioblastoma multiforme (GBM) is one of the most aggressive human cancer, characterized by high invasiveness and resistance to the current standard care [1]. Despite the progress in the therapy of this tumor, consisting in maximal safe surgical resection followed by radiotherapy and temozolomide (TMZ), the prognosis of patients suffering from GBM remains poor, with a median survival of only 1.5 years [2].
A number of papers pointed out that tumor relapse could be due to a restrict population of cells, endowed with tumor initiating potential and resistance to radiochemotherapy [3, 4], which are commonly referred to as glioma stem-like cells (GSCs). Indeed, they share some properties with the neural stem cells (NSCs), resident in specific zones of the adult brain [5–7]. Moreover, it has been suggested that GSCs could arise from the malignant transformation of NSCs, mainly deriving from the subventricular zone, from which they would migrate leading to the development of glioma in different cerebral areas [4, 8].
GSCs have been isolated from GBMs and grown as neurospheres in vitro, showing a high capacity for self-renewal and aberrant differentiation. Additionally, these cells are able to form tumors after grafting in immunosuppressed mice [9, 10], demonstrating that they are required for tumor development and maintenance [11]. Although the definitive role of GSCs in GBM ontology and recurrence remains elusive, these properties attracted a great interest by researchers and clinicians as new targets of the therapy of this tumor.
Extracellular purine nucleosides (adenosine, guanosine) and nucleotides (mainly ATP and GTP) have long been recognized as modulators of neurotransmission and trophic factor production in the adult central nervous system (CNS), where they contribute to regulate neuronal plasticity and glial function [12]. Accordingly, they have also been implicated in the pathophysiology of numerous pathological conditions ranging from brain trauma and ischemia to neurodegenerative and neuropsychiatric disorders [13]. More recently, purinergic signaling has been shown to be involved in embryological/postnatal brain development [14, 15] and NSC activities [16–20] and to play a role in cancer biology [21]. In this regard, some reports on purine and pyrimidine analogs, used as antileukemic agents or experimental cytotoxics to treat solid tumors [22–25], showed that they behave as antimetabolites competing with physiologic nucleosides [26]. In contrast, many other in vitro and in vivo studies on different cancer types including brain tumors demonstrated the antitumoral activity of purines as being linked to the selective stimulation of specific purinoceptors [21, 27–33]. While only brain-specific binding sites for guanine-based compounds have been discovered [34], receptors for adenine-based purines are found in all neural cells [35]. They are divided into P1 (P1R) and P2 (P2R) receptors recognizing adenosine and ATP as the main ligands, respectively [36]. P1R family comprises 4 subtypes, namely A1R, A2AR, A2BR, and A3R, whereas P2R family is subdivided into seven ionotropic P2XR and eight metabotropic P2YR, which have currently been cloned in mammalian species and characterized [37, 38]. It is to highlight that studies on purine effects are in general complicated by the presence of either P1R or P2R on the same cell, often playing opposite roles, and by the existence of ectoenzymes rapidly transforming nucleotides into active nucleosides [39].
In the last 10 years, data have been published on the activity of extracellular purines, mainly adenine-based nucleotides, acting as antitumoral drugs in human and rat brain tumor cell lines [30, 33, 40]. However, the relevance of conventional cell lines (cultured under mainly serum-based media conditions) as means to investigate GSCs is disputed, as the results may be biased by multiple new mutations induced during long-term culture in serum-containing media. On the contrary, GBM cell lines derived from freshly resected tumor specimens and cultured in serum-free medium supplemented with EGF and bFGF—conditions optimized for the growth of neural stem cells—mirror the phenotype and genotype of primary tumors more closely than serum-cultured cell lines do [41, 42]. Therefore, since GBM stem cell cultures may be a more reliable model to appreciate the efficacy of therapeutic agents, we thought to reinvestigate the anticancer effects of purine compounds (natural and synthetic ligands) on these cells, checking whether they acted at extracellular levels as signaling molecules, as previously shown in conventional cell lines so far studied [30, 40, 43]. Additionally, in order to make our experimental contribution potentially useful for the improvement of the current GBM therapy, we evaluated whether purine nucleotides might increase the toxicity of TMZ and the duration of its effect on these cells.
Methods
Chemicals
3-[[5-(2,3-Dichlorophenyl)-1H-tetrazol-1-yl]methyl]pyridine hydrochloride (A438079), 3-[1-[[(3′-nitro[1,1′-biphenyl]-4-yl)oxy]methyl]-3-(4-pyridinyl)propyl]-2,4-thiazolidinedione (AZ 11645373), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), N-[9-Chloro-2-(2-furanyl) [1, 2, 4]-triazolo[1,5-c]quinazolin-5-yl]benzene acetamide (MRS1220), [[(1R,2R,3S,4R, 5S)-4-[6-Amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl]diphos-phoric acid mono ester (MRS2365) trisodium salt, (1R*,2S*)-4-[2-iodo-6-(methyl amino)-9H-purin-9-yl]-2-(phosphonooxy)bicyclo[3.1.0]hexane-1-methanol-dihydrogen-pho-sphate ester (MRS2500) tetra-ammonium salt, 4,4′-(Carbonylbis(imino-3,1-(4-methyl-phenylene)carbo-nylimino))bis(naphthalene-2,6-disulfonic acid) tetrasodium salt (NF340), 4,4′-(Carbonylbis (imino-3,1-phenylene-carbonylimino-3,1-(4-methyl-phenylene)carbonylimino))-bis(1,3-xylene-alpha,alpha′-diphosphonic acid tetra-sodium salt (NF546), 8-[4-[4-(4-chlorophenzyl) piperazide-1-sulfonyl)phenyl]]-1-propylxanthine (PSB603), and 4-(2-[7-amino-2-(2-furyl) [1, 2, 4]triazolo[2,3-a][1, 3, 5]triazin-5-ylamino]ethyl)phenol (ZM241385) were purchased from Tocris (Space Import/Export, Milan, Italy); apyrase (VII grade), 2′(3′)-O-(4-benzoylbenzoyl)-ATP (Bz-ATP), periodate-oxidized-ATP (oATP), suramin, and TMZ were from Sigma-Aldrich as well as all other chemicals, unless differently specified (Milan, Italy).
Cell cultures
We used GSCs obtained from three different patients with a primary tumor, the molecular profile of which is reported in Table 1. Once isolated, cells were grown in serum-free medium, supplemented with mitogens (20 ng/ml of human recombinant EGF and 10 ng/ml of human recombinant bFGF), as previously described [44, 45]. Under these culture conditions, tumor-derived cells formed classical floating aggregates called tumor neurospheres, which were used for some experiments. However, whereas the most part of them were performed on cells grown as monolayer obtained precoating culture plates with Matrigel (BD Bioscience, Buccinasco, Italy) dissolved in culture medium and then seeding the cells that were fed with the usual culture medium containing also the growth factors above mentioned (dilution 1:200). In this condition, GSCs maintained spherogenic properties [44, 45], but their use allowed a more precise quantification of cell survival in vitro in comparison with neurospheres. It is to underline that cells used in this study were previously characterized for many biologic/molecular features including the retention of stemness markers, the resistance to chemotherapeutic agents, the ability of self-renewing in vitro or generating a tumor identical to the human tumor in antigen expression and histological tissue organization when injected in the brain of immunocompromised mice [44–46].
Experimental protocols
Using undifferentiated GSCs grown as either monolayer or neurosphere suspension, purinergic drugs and also apyrase, the enzyme catalyzing the hydrolysis of ATP to yield AMP, were administered on the 2nd day after cell plating and renewed on the next day, whereas TMZ, either alone or in combination with purine receptor ligands, was usually administered only once, on the 2nd day after cell plating, except in the last experiment (see Fig. 7) in which it was administered a second time alone, 4 days after the first treatment. Purine receptor antagonists, when present, were administered to the cultures 30 min prior to the agonists. In the experiments with undifferentiated GSC monolayers, cell viability or apoptotic/necrotic death was measured on untreated/treated cells on the fourth and also on the seventh day after seeding. In the experiments with neurospheres in suspension, the effect of the pharmacological treatments was estimated at different intervals from the drug administration as the capability of the drugs to affect the process of neurosphere formation. The neurosphere size was evaluated the first time at day 7 after the pharmacological treatment. Then, the effect of the same pharmacological treatment was assessed on the GSC ability to form secondary neurospheres from the primary ones, which is usually considered as an index of tumor invasiveness. In this kind of experiments, untreated/treated neurospheres were allowed to recover for further 7 days (14 days from the beginning of the experiment); then, they were removed, centrifuged, mechanically dissociated, and resuspended at fixed number (10,000/well). The formation of secondary neurospheres was assessed at day 21 from the beginning of the experiment.
Analysis of CD39 expression, cell cycle distribution, and apoptosis by flow cytometry
To analyze the expression of CD39, a typical cell surface protein marker, 5 × 105 cells/sample, was incubated with 100 μl of 20 mM ethylenediaminetetraacetic acid (EDTA) at 37 °C for 10 min. Cells were washed with 3 ml of washing buffer and centrifuged (4 °C, 400 × g, 8 min). Subsequently, samples were resuspended in 100-μl washing buffer containing the appropriate amount of fluorescein isothiocyanate-conjugated (FITC antibody against CD39 (Becton Dickinson, BD, San Jose, CA)) and incubated for 30 min at 4 °C in the dark. Tubes were washed (3 ml of washing buffer), centrifuged (4 °C, 400 × g, 8 min), and cells were resuspended with 1-ml 0.5 % paraformaldehyde, incubated for 5 min at room temperature, washed, centrifuged (4 °C, 400 × g, 8 min), and stored at 4 °C in the dark until the acquisition with a FACSCalibur flow cytometer (BD), using the CellQuest™ 3.2.1.f1 software (BD). Ten minutes before the analysis, a specific solution, containing 7-amino-actinomycin D (BD), was added to the samples to exclude dead cells. Quality control was performed by a regular check-up with Rainbow Calibration Particles (BD Biosciences). Debris was excluded from the analysis by gating on morphological parameters; 20,000 nondebris events in the morphological gate were recorded for each sample. Data were analyzed using the FlowJo™ software (TreeStar, Ashland, OR). Mean fluorescence intensity ratio (MFI Ratio) was calculated dividing the MFI of positive events by the MFI of negative events.
To assess cell cycle distribution or apoptosis, 5 × 105 cells/6-well were exposed to various concentrations of purinergic ligands for 48 h. Cells were collected, and cell cycle distribution was analyzed by propidium iodide (PI) flow cytometry. Both floating and attached cells were collected and poured together into centrifuge tubes. They were rinsed with PBS and fixed with 70 % ice-cold ethanol containing 2 mg/ml RNase for 30 min. Subsequently, cells were washed 2 times with PBS and ultimately stained with PI (40 μg/ml) at room temperature for 10 min. PI fluorescence of cells was determined by the same Flow Cytometer (BD Biosciences) with excitation wavelength of 488 nm and emission of longer than 590 nm. The propidium iodide fluorescence of 20,000 cells was counted for the control and purine agonist-treated samples. The percentage of cells in G0/G1 phase, S phase, and G2/M phase was calculated by use of standard ModiFit and CellQuest software programs [47].
Quantification of apoptosis by caspase 3/7 activity
Quantification of apoptosis in GSCs, treated with appropriate concentration of P2 agonist receptors, was performed using Caspase-Glo Assay Tecnology by providing a luminogenic caspase 3/7 substrate, which contains the tetrapeptide sequence DEVD, in a reagent optimized for caspase acitivty, the luciferase activity. Luciferase activity is proportional to the amount of caspase activity present. The assay was carried out according to the instructions of the supplier company (Promega Italia, Milan, Italy).
RNA isolation and RT-PCR analysis
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s recommendations. The resulting RNA pellet was dissolved in 30-μl diethyl-pyrocarbonate-treated water, and 5 μg was run on formaldehyde denaturing gel to confirm the integrity of the RNA. To remove any genomic DNA contaminants, RNA samples (10 μg) were treated with 1 U DNase-I RNase-free (Roche, Monza, Italy). First, strand cDNA was synthesized from 1.5 μg of total RNA using the RT-PCR system RETROscript™ (Ambion, Monza, Italy) with random hexamers. The resultant cDNA (2 μl) was amplified in a 100-μl reaction volume containing PCR reaction buffer, 1.5 mM MgCl2, 0.2 mM each deoxy-dNTP, 1 μM oligonucleotide primers (MWG Biotech, Ebersberg, Germany), 2.5 U AmpliTaq Gold™ DNA polymerase (Applied Biosystems, Life Technologies). The final cycle was followed by a 10-min incubation at 72 °C. All primers were synthesized by MWG Biotech. PCR primers, annealing temperatures, and product sizes are shown in Table 2. Reaction was also performed without the reverse transcriptase step as control for genomic contamination. PCR products were separated by 1.5 % agarose gel electrophoresis in gels containing ethidium bromide and visualized with UV light.
Western blot analysis
Cells were harvested at 4 °C in a lysis buffer (in mM: Tris buffer 50, NaCl 150, PMSF 1.0; 1 % Nonidet-P40, 5 μg/ml leupeptin, 5 μg/ml aprotinin), disrupted by sonication and centrifuged (14,000 rpm, 5 min, 4 °C). Protein concentration was determined by BioRad protein assay (Bio-Rad Laboratories, Milan, Italy). Samples (50 μg), diluted in SDS-bromophenol blue buffer, were boiled (5 min) and separated on 12.5–15 % SDS-polyacrylamide gels. Proteins were transferred on a polyvinylidene fluoride membrane, blocked with PBS/0.1 % Tween20/5 % nonfat milk (Bio-Rad Laboratories) for 2 h at 4 °C, incubated overnight at 4 °C with specific primary antibodies [polyclonal rabbit anti-A1, anti A2A anti-A2B, anti-A3, anti P2Y1-2-4-6-11-12-13-14, anti-P2X7, dilution 1:1000 (Alomone Labs, Jerusalem, Israel)]; polyclonal rabbit anti CD73, dilution 1:500 (Novus Biological Europe, Cambridge, UK) and then exposed to donkey anti-rabbit HPR-conjugated secondary antibody for 1 h at room temperature (GE Healthcare Life Sciences, Milan, Italy; final dilution 1:2500). The specificity of the antibodies used to determine the presence of adenosine receptors was previously established [48–51]. To determine the equal loading of samples, the blots were stripped and reprobed with an anti-ß-actin antibody (dilution 1:1000, incubation overnight at 4 °C; Santa Cruz Biotechnologies). Immunocomplexes were visualized using the enhancing chemiluminescence (ECL) detection system (GE Healthcare Life Sciences) and quantified by densitometric analysis (Molecular Analyst System).
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) levels are widely used to estimate necrotic cell death since LDH is a cytoplasm enzyme that can be released following cell membrane damages. Cells, seeded (3 × 103 cells/well) in 96-well plates, were incubated with agonists of P1 and P2 receptors following the usual protocol. Twenty-four hours after the end of the treatment, the cells were incubated at 37 °C and 5 % CO2 for 45 min with specific lysis buffer, and then, the plate was centrifuged at 250 g for 4 min. Then, 50 μl of supernatant from each well, transferred to a new 96-well plate, was added to 50 μl of substrate buffer consisting of 0.7 mM p-iodonitrotetrazolium Violet, 50 mM L-lactic acid, 0.3 mM phenazine methoxysulfate, 0.4 mM NAD, and 0.2 M Tris-HCl pH 8.0. The plate suitably blanket was incubated in the dark at room temperature for 30 min, and finally, the reaction was stopped by addition of 50 μl/well of stop solution. The absorbance was measured spectrophotometrically at 490 nm, and the results were expressed as a percentage of total LDH released from the positive control consisting of cells treated with 25 μl of 10 % Nonidet P-40 (NP-40) and calculated as follows: (supernatant absorbance value − white absorbance value) / (supernatant absorbance + lysate absorbance) × 100. All reagents were from Promega Italia (Milan, Italy).
Cell proliferation
Cell proliferation was assayed by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay, using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega Italia, Milan, Italy), according to the manufacturer’s instructions. The absorbance was measured at 490 nm, using a microtiter plate reader (Spectracount™, PerkinElmer Life, Waltham, MS, USA).
Statistical analysis
All results are presented as mean ± SEM. Data were analyzed by the Student’s t test, with a significance level of 95 %. For the analysis of difference between more than two groups, one-way analysis of variance (ANOVA) was used followed by Dunnett’s post hoc test. Difference was considered to be statistically significant at a value of p < 0.05. Analyses were performed with GraphPad Prism 4 (GraphPad, San Diego, CA, USA).
Results
GSC line characterization
We used GSCs deriving from samples of primary GBMs obtained from three patients. The primary tumors showed various molecular profiles (Table 1), which could be correlated with the patient outcome and the response to TMZ in malignant gliomas [52, 53]. Notably, tumor from the patient no. 3, differently from those of the other two patients, showed an unmethylated status of the enzyme O6-methylguanine methyltransferase (MGMT) promoter compatible with the shortest period of overall survival of that patient, as reported elsewhere [54].
Following standard protocols, GSCs were cultured in the presence of specific growth factors (see “Methods” section) either as cell suspension, which allows the formation of the classical neurospheres, or as adherent cells on a matrigel substrate (Fig. 1a). In both cases, GSCs maintained an undifferentiated state, as indicated by cell morphology.

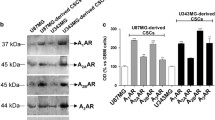
Human GSCs express different subtypes of purine receptors and two stemness markers such as CD39 and CD73, corresponding to the enzymes ectonucleoside triphosphate diphosphohydrolase 1 and 5′-ectonucleotidase. a GSCs, isolated from human primary GBM and used for experiments, were cultured either as cell suspension leading to the formation of classical neurosperes or as cell monolayer when seeded on plates coated with a suitable substrate (Matrigel). The panels show cultures obtained from the patient no. 3 and are representative of similar cultures obtained from the other two patients selected for this study. b–e The presence of metabotropic receptors belonging to the P1 family (d), responsive to adenosine, and to the P2 family (e) responsive to ATP/ADP (P2Y1,11-14) and UTP (P2Y2,4,6) as well as the expression of the ionotropic P2X7 receptor (e) and of CD73 (b) have been evaluated by either RT-PCR or Western blot analysis whereas the expression of CD39 was assayed by cytofluorimetric analysis (c). Bands or histograms were obtained from GSCs deriving from surgical tissue samples of three different patients identified as # 1, 2, and 3. In the c, a semiquantitative evaluation of antigen expression, obtained dividing the mean fluorescence ratio (MFI) of positive events by the MFI of negative events, is reported. In b–e, for RT-PCR assay, a lane is reported on the left of each panel in which standard molecules (M) were run, and within brackets, it has been reported the indication of the base pairs (bp) of the products that are those currently indicated in literature for each receptor subtype. Expression of GAPDH or β-actin was used as an internal control. The bands obtained by Western blotting were analyzed by densitometry, and the obtained values were normalized for those evaluated for β-actin bands and are reported in the tables. Data are representative of three independent experiments with very similar results
GSC express P1 and P2 purinoceptors
As first step, we ascertained the expression of P1R and P2R in GSCs, focusing on the metabotropic ones. We also looked for ionotropic P2X7R, which is present in glioma cell lines [43] and causes death in a number of cells [55, 56]. By either RT-PCR or Western blot techniques, the presence of all four adenosine receptors was detected in GSCs, with the A2BR protein expressed to the lowest extent (Fig. 1d). In the same cells, all P2YR responsive to ATP were present, whereas, among those responsive to pyrimidine nucleotides, P2Y2R showed the greatest expression (Fig. 1e). Additionally, GSCs expressed high levels of P2X7R (Fig. 1e). We also ascertained the presence of the stem cell markers CD39 and CD73, corresponding to the enzymes ectonucleoside triphosphate diphosphohydrolase 1 and 5′-ectonucleotidase (Fig. 1b, c), respectively, the activity of which is fundamental for the metabolism of extracellular nucleotides [43].
P2 receptor agonists significantly reduced GSC growth
Initially, the effect due to extracellular ATP, present as nutrient or released from cells in the culture medium, was assayed exposing cells to apyrase, which metabolizes ATP and ADP, therefore reducing the amount of extracellular active nucleotides. Apyrase alone did not modify cell growth, as shown in Fig. 2a where we reported the effect measured in GSCs from the patient no. 3, for which the results were similar to those obtained from cells of the other two patients (data not shown).

Effect of purine receptor agonists and apyrase on the GSC viability. Undifferentiated cells grown on Matrigel were exposed for two consecutive days to: a apyrase, administered alone or in combination with different doses of Bz-ATP. Values are the mean ± SEM of 3 separate experiments using cells from the patient # 3. Similar values were obtained using GSCs from the other two patients. *p < 0.05, ***p < 0.001: statistical significance vs control cells; §§ p < 0.01, §§§ p < 0.001: statistical significance vs. cells exposed to Bz-ATP alone (one-way ANOVA plus Dunnett’s test); b different doses of nonselective agonists for P2 receptors (ATP and UTP) or of more selective agonists for P2Y1 (MRS2365 and ADPβS), P2Y11 (NF546), and P2X7 (Bz-ATP) receptor subtypes. Data reported in b are representative of six independent experiments, carried out using cells derived from three different patients. In all figures, cell viability evaluated by the MTS assay as the absorbance detected at 490 nm was measured after two further days following cell exposure to drugs
Then, the effects of the stimulation of different P2R subtypes were assayed on the proliferation of GSCs, using natural (ATP and UTP, both nonselective P2R agonists), and synthetic ligands (MRS2365 and NF546, currently the most selective agonists of P2Y1R and P2Y11R, respectively, and Bz-ATP, P2X7R agonist). We also evaluated the effect of ADPβS, a less selective agonist for P2Y1R in comparison to MRS2365, which may also interact with P2Y12-13R. All agents were administered in a range of concentrations from 1 μM up to 1 mM, except MRS2365, employed at concentrations from 1 nM to 10 μM.
MRS2365 and, to a lesser extent, ADPβS increased cell proliferation rate in dose-dependent fashion, most in GSCs from the patient no. 3. The same did ATP, stimulating cell duplication up to the dose of 150 μM (Fig. 2b). In contrast, when administered at greater concentrations, ATP inhibited GSC proliferation in dose-dependent manner and Bz-ATP, starting from the concentration of 100 μM onwards, did the same whereas UTP reduced cell viability only at the highest doses (500–1000 μM) and NF546 did not affect it at all (Fig. 2b). The GSCs isolated from the patient no. 3 showed a lower sensitivity to ATP or Bz-ATP cytotoxicity. Interestingly, the presence of apyrase together with submaximal doses of Bz-ATP further reduced cell viability, and this effect was significant even in less responsive GSCs from patient no. 3 (Fig. 2a).
The effect of P2YR and P2X7R agonists on the growth of GSCs was counteracted by the respective antagonists
We investigated whether the effects caused by purine nucleotides were mediated by the interaction with P2R, using the antagonists commercially available. All compounds, when administered alone, did not affect cell viability, except o-ATP, an older P2X7R antagonist (Fig. 3a), which was no longer used in the subsequent experiments. In the presence of purinergic agonists, MRS2500, P2Y1R selective antagonist, reduced the proliferative effect of MRS2365, ADPβS, and ATP (Fig. 3b, c). On the other hand, the antiproliferative effect of a submaximal dose (500 μM) of ATP was in part reversed by either suramin, nonselective P2R antagonist, or AZ11645373, but not by TNP-ATP, which blocks all P2XR except the P2X7 one, or by NF340, blocking the P2Y11R (Fig. 3b). AZ11645373 (Fig. 3c) and A438079 (data not shown) also inhibited the effect of low (100 μM) but not of high (500 μM) doses of BzATP, whereas suramin did not counteract the effect of 500 μM UTP. As expected, NF340 did not alter GSC growth in the presence of the P2Y11R agonist NF546 (Fig. 3c).

Effect of nucleotides, administered alone or in the presence of different purine receptor antagonists, on the growth of GSCs. a Cells grown on Matrigel were exposed for two consecutive days to selective antagonists of P2Y1R (MRS2500), P2Y11R (NF340), and P2X7R (A438079, AZ116453, o-ATP) and to nonselective antagonists of P2R (suramin) and P2XR (TNP-ATP). b, c The effect caused by cell exposure for two consecutive days to selective agonists for P2Y1R (MRS2365 and ADPβS) and P2X7R (Bz-ATP) or to nonselective agonists (ATP or UTP) for P2 receptors, was evaluated in GSCs, in the presence or not of the antagonists reported above. When present, the antagonists were administered 30 min prior to the agonists. Cell viability evaluated by the MTS assay as the absorbance detected at 490 nm was measured after two further days following cell exposure to drugs. Values are the mean ± SEM of 6 separate experiments in which different cell samples were used. *p < 0.05, *** p < 0.001: statistical significance vs. control cells; § p < 0.05, §§§ p < 0.001: statistical significance vs. cells exposed to purine nucleotides alone (one-way ANOVA plus Dunnett’s test)
Inhibitory effect of adenosine on the growth of GSCs
Next, the activity of nucleosides, such as adenosine, inosine, and uridine, again administered in the range from 1 μM to 1 mM for two consecutive days, was assayed to verify if they might be responsible for the effect caused by purine/pyrimidine nucleotides. Only adenosine, at the highest doses, reduced the cell survival by about 40–60 % (Table 3), whereas the other assayed nucleosides did not significantly affect GSC growth (data not shown). To prove the involvement of one or more adenosine receptors (AR), we tested the adenosine effect in the presence of DPCPX (100 nM), ZM241385 (100 nM), PSB603 (300 nM), and MRS1220 (1 μM), selective antagonists of A1R, A2AR, A2BR, and A3R, respectively. Among these, MRS1220 (Table 3), but not the other compounds (data not shown), counteracted the adenosine effect. MRS1220 was also able to limit the inhibitory activity of ATP high doses on cell growth (Table 3), thus suggesting that the nucleotide effect could in part be mediated by the formation of extracellular adenosine that in turn, interacting with A3R receptor, may cause cell arrest.
Synergistic antitumoral effect of the combined treatment of GSCs with TMZ, a classic antitumoral agent for GBM therapy, and purinoceptor ligands
In another set of experiments, we evaluated the antiproliferative activity of purine compounds in the presence of TMZ, an agent currently used in the chemotherapy of GBM [2]. TMZ, administered alone to the cultures (Fig. 4a), caused a dose-dependent reduction of cell survival, which was still evident after 7 days. When it was administered at 50 and 75 μM in the presence of submaximal doses of ATP or UTP or BzATP, the antiproliferative effect of these nucleotides was significantly enhanced (Fig. 4b), except in the GSCs from the patient no. 3, less sensitive to the cytotoxicity induced by all compounds.

Effect of temozolomide (TMZ), administered alone or in combination with purine receptor agonists on the growth of GSCs. a Undifferentiated GSCs derived from samples of different patients and grown on Matrigel were exposed to TMZ, administered only once at different concentrations on the 2nd day after cell seeding. Cell viability, evaluated by MTS assay as the absorbance detected at 490 nm in untreated cultures (control, CTRL) and in cultures exposed to TMZ, was detected at 4 and 7 days from the beginning of the experiments. The results are expressed as the mean ± S.E.M. of three independent experiments, in which different cell samples were used and evaluated in triplicate; b GSCs derived from the same three patients and grown on Matrigel, were exposed for two consecutive days to 500 μM ATP or UTP, nonselective agonists for P2 receptors, or Bz-ATP, selective agonist for P2X7R, alone or in combination with to 50 or 75 μM TMZ, administered once on the second day together with purine/pyrimidine nucleotides. Cell viability, evaluated again by MTS assay as the absorbance detected at 490 nm, was measured at the day 4 from the beginning of the experiments. *p < 0.05, ***p < 0.001: statistical significance of vs. untreated (control) cells; § p < 0.05, §§§ p < 0.001: statistical significance vs. cells exposed to TMZ alone (one-way ANOVA plus Dunnett’s test)
We wondered whether the reduction of cell viability by purine compounds, alone or combined with TMZ, was due to cell death or cell growth impairment. All assayed nucleotides significantly increased cell necrosis as revealed by the measurement of LDH release (Fig. 5a); ATP and UTP remarkably enhanced also the cell apoptotic death as pointed out by the assay for the caspase 3/7 levels and/or by the propidium iodide assay (Fig. 5b, c). Additionally, ATP diminished the number of cells in the G2/M phase, whereas Bz-ATP accumulated them in the S phase (Table 4). Thus, the decrease in cell viability was in part due to a cytotoxic effect by purine nucleotides, which caused also an arrest of cell proliferation acting at intracellular level, likely on DNA duplication process.

Evaluation of cell necrotic or apoptotic death as a consequence of GSC exposure to purine/pyrimidine nucleotides or TMZ, administered alone or in combination. The effect caused by cell exposure for two consecutive days to purine nucleotides (ATP, UTP, or BzATP) and for 1 day to TMZ, administered alone or in combination (see the experimental protocols reported in the text of the paper) was evaluated in GSCs from the patient no. 3. Similar values were obtained using GSCs from the other two patients. a LDH release from GSCs, assumed as an index of necrotic death, was measured as reported in the “Methods” section. Values are expressed as the percentage of the total amount of the enzyme released in the medium from the cells after their lysis and are the mean ± SEM of 6 separate experiments in triplicate. b, c Apoptic death was assessed by two methods. The first one evaluated the release of caspase 3 and 7, the most involved in this process, by luminescence using a commercial kit and following the manufacturer’s instruction. Cell apoptosis was also measured by incubating cells with propidium iodide and revealing the incorporated fluorescence by FACS. The values in the b and c are the mean ± S.E.M. of four independent experiments in which each sample was tested in triplicate. *p < 0.05, ***p < 0.001: statistical significance vs. untreated cells; §§ p < 0.01, §§§ p < 0.001: statistical significance vs. cells treated with TMZ (one-way ANOVA plus Dunnett’s test)
Also, the inhibitory effect of TMZ alone on GSC growth was due in part to the increase of the percentage of either apoptotic or necrotic cells and to a substantial arrest of the cell growth with a consequent increase of their number in the S and G2/M phases of the cell cycle (Table 4 and Fig. 5b, c). When TMZ was administered together with ATP, the number of apoptotic cells significantly increased, even though there was no evident involvement of the caspase 3/7 activity (Fig. 5b, c). Moreover, this combined treatment further enhanced the percentage of GSCs arrested in the G2/M phase of the cell cycle, although this effect did not reach a statistical significance when compared to the effect of TMZ alone (Table 4). In contrast, the contemporaneous presence of Bz-ATP with TMZ determined a more relevant necrotic process (Fig. 5a), whereas the changes in the amount of GSCs in the G2/M phase or in the S phase of the cell cycle were equivalent to the effect of Bz-ATP alone, the effect of which was prevalent over that exerted by TMZ alone (Table 4).
Since GSCs are provided with many mechanisms for DNA repair or against apoptosis [57], we checked whether the effect caused by nucleotides in combination or not with TMZ on cell growth was long-lasting. As expected, cell proliferation evaluated by MTS assay was restored 14 days after the pharmacological treatment (data not shown).
Effect of purine/pyrimidine compounds and TMZ on the formation of secondary neurospheres following different period from the initial treatment of primary neurospheres
We tested the effect of purine/pyrimidine nucleotides and/or TMZ, added to the cells following the usual protocol, on the formation of neurospheres that develops when GSCs are cultured as cell suspension and could result more resistant to any pharmacological treatment. The size of surviving neurospheres, measured 7 days from the beginning of the experiment, was significantly reduced by cell exposure to nucleotides (Fig. 6a–c). In contrast, the size of secondary neurospheres, considered as an index of tumor invasiveness, was similar to that of control, except for GSCs exposed to Bz-ATP or TMZ alone, which significantly reduced it. The administration of TMZ in the presence of purine/pyrimidine nucleotides did not cause further significant reduction in the size of primary or secondary neurospheres, measured after 7 or 21 days, respectively, in comparison to the effect of nucleotides alone (Fig. 6b–d).

Evaluation of the effect of purine receptor agonists on the formation of neurospheres by GSCs. GSCs grown as cell suspension were exposed for two consecutive days to ATP or UTP or Bz-ATP or TMZ, administered alone or in combination as reported in the experimental protocol. a The formation of neurospheres was first evaluated after 7 days from the cell exposure to the drugs. Then, the cells were allowed to recover for further 7 days and, subsequently, after a recovery period of 7 days, the surviving neurospheres were submitted to dissociation. b Cells obtained from this procedure were reseeded and evaluated after further 7 days following dissociation for their ability to form secondary neurospheres. The panels a–b are related to experiments performed using cells from the patient no. 3 and are representative of six independent experiments performed using GSC deriving from different patients and which gave similar results. The cells were observed with a Nikon Eclipse TS100 phase contrast microscope, and images were acquired with the Zoom Browser EX software. Original magnification, ×20. In the graphs below the panels (c, d), values related to the size of primary and secondary neurospheres are reported. They were obtained using cells from three patients. The values are the mean ± S.E.M. of three independent experiments in which each sample was tested in triplicate. *p < 0.05, **p < 0.01: statistical significance vs. untreated cells (control) (one-way ANOVA plus Dunnett’s test)
As the last experiment, we assayed a further experimental protocol, administering TMZ alone a second time, which is 4 days after the initial treatment performed with TMZ in combination with nucleotides. Also, in this case, the effect of TMZ was greater in cells previously treated also with ATP and Bz-ATP, but the effect was still evident after 14 days even in GSCs from the patient no. 3 (Fig. 7).

Effect of TMZ, administered twice (the first time in combination with purine receptor agonists and the second time alone) on the growth of GSCs. Using GSCs grown on Matrigel, cells were exposed for two consecutive days to 500 μM ATP or UTP or BZ-ATP and to 50 or 75 μM TMZ, administered on the second day together with purine nucleotides. After three further days, cells were reexposed to TMZ alone. Cell viability was detected at the day 14 from the beginning of the experiments. Values reported in the graph are the mean ± SEM of 6 separate experiments in triplicate using cells from all patients. ***p < 0.001: statistical significance of vs. untreated (control) cells; §§§p < 0.001: statistical significance vs. cells exposed to purine nucleotides alone (one-way ANOVA plus Dunnett’s test)
Discussion
GBM is the most common and aggressive primary tumor of the adult CNS, against which the current therapy is largely ineffective [1, 2]. In the attempt to find novel druggable targets to combat this tumor, we investigated the antitumoral effects of purine/pyrimidine nucleotides that are able to interact with specific own receptors. These agents were administered alone or in combination with a specific antitumoral agent, the TMZ, in GSCs deriving from primary human tumors, which are believed to be responsible of GBM relapse/invasiveness [3, 4]. We had the opportunity to use cells isolated from primary tumors with different biological characteristics and various expression of molecular prognosticators (Table 1) [52–54]. Interestingly, GSCs from the tumor of the patient no. 3, who had a shorter period of overall survival in comparison with the other two patients, showed a decreased response to the antiproliferative effects induced by TMZ and also by purine nucleotides. It is reasonable to hypothesize that this could be due to intrinsic mechanisms of greater resistance of these cells (i.e., processes related to DNA repair) or to a dysfunction of some of the investigated receptors and not to differences in their expression, which was similar in all three kinds of cells, in agreement with levels reported for glioma cell lines [43, 58]. Of course, this aspect should deserve a deeper investigation.
Noteworthy, the expression of P2X7R in our GSCs resulted more pronounced than that of other purinergic receptors. In agreement with these data, we here demonstrated that P2X7R plays a pivot role in inhibiting GSC growth. In fact, our cells were sensitive to the cytotoxic effects caused by Bz-ATP, P2X7R agonist, even when it was administered to GSCs grown as neurospheres. A similar effect was found also in cells exposed to high concentrations of ATP, which are known to stimulate the same receptor. Finally, Bz-ATP and ATP effects were in part counteracted by the P2X7 antagonist. Similar findings were reported in glioma cell lines in which a toxic effect was induced by purine nucleotides coupled to a prevalent involvement of P2X7R [reviewed in reference 43], whereas our results differ from those obtained by Ledur et al. [40], who showed a toxic effect of ATP low concentrations on the growth of tumor spheres obtained from those cell lines, especially in the absence of serum supply. We hypothesize that this difference may be due to a greater resistance of GSCs deriving from freshly resected human glioblastoma to nucleotide toxic insults.
ATP antitumor activity, in part likely due to its conversion into adenosine by ectoenzymes present in the GCS membrane, was reduced also by cell pretreatment with MRS1220, an A3R antagonist that restrained also the citotoxicity caused by high doses of adenosine. The A3R anticancer effect, like that of P2X7R, has been described in other malignancies [59–62], even though other papers argued the opposite [63–66]. Of note, there was no involvement in the control of GSC growth by other AR, such as A1R and A2BR, the stimulation of which caused a prominent antiproliferative effect on stem cells from human GBM cell lines [67]. Such a discrepancy may depend on differences in receptor expression and function in tumor cells, in turn depending also on the source of GSCs, which in our case derive from primary tumors.
Our findings indicate that purine effects are only in part receptor-mediated. Indeed, the cell cycle analysis suggested that GSC growth impairment was due also to their direct interference with DNA duplication, as previously reported [26]. Also, this aspect should be taken into due consideration for future clinical application of these compounds, as the failure of cell duplication in normal tissues might cause possible broader side effects. So far, ATP has been used as a compassionate drug in terminally ill patients [68, 69]. The systemic administration did not cause important side effects indicating that the therapy with this compound may be tolerated [70]. Nothing is known as for the administration of Bz-ATP or other compounds acting on P2X7R in humans, and the investigation in animals is just at the beginning [71, 72].
Noteworthy, ATP may also favor GSC proliferation. The trophic role of ATP, reported by other authors in human glioma cell lines [30], is also suggested by the effect of apyrase, able to metabolize extracellulare ATP, thus reducing its presence in the growth medium. In this way, although ineffective when administered alone, apyrase likely potentiated the inhibitory activity of moderate doses of the synthetic P2X7R agonist, Bz-ATP, on CGS duplication. P2Y1R are involved in the ATP-induced tumor growth as MRS2365 and ADPβS, P2Y1R agonists, showed an effect similar to that of a low dose of ATP whereas MRS2500, P2Y1R specific antagonist, counteracted the trophic effect of all agonists cited above. A tumorigenic role of P2Y1R was reported for embryonic carcinoma cells [32], in which, differently from our findings, the stimulatory effect on cell growth was shared with the activation of P2Y2R, responsive to UTP. However, in other tumor cells, i.e., in prostate cancer cells, P2Y1R stimulation caused apoptosis [73].
Other ATP P2 receptors do not seem to be involved in the control of GSC proliferation. In fact, TNP-ATP, able to block all P2XR except P2X7R, did not counteract ATP effects; also, the involvement of P2Y11R could also be excluded as well as that of P2Y12-13R, as the proliferative effect of ADPβS, agonist also for these receptors, was completely reversed by the P2Y1R antagonist. These findings are different from that obtained in murine and human cell lines, in which P2Y12R appear to increase cell proliferation, whereas P2Y1R have opposite effects in different cell lines, causing either an increase or a decrease in the cell population [43]. Altogether, our results confirm the need to evaluate the pattern of purinoceptor expression and function, which is specific for each tumor.
Finally, in our opinion, the effects obtained by the cell exposure to TMZ in combination with cytotoxic doses of purinergic compounds are remarkable. For the first time, we demonstrated that this combined pharmacological treatment resulted in a potentiated antiproliferative effect, at least in cells derived from primary GBMs grown as monolayer, which likely were more easily accessible by the drugs. In particular, cell exposure to ATP or BzATP plus TMZ significantly enhanced the percentage of apoptotic or necrotic cells. Apart a possible increased cytotoxicity induced by the nucleotides, a tentative explanation of the potentiated effect might be that ATP or BzATP, by opening the pore associated to the P2X7R, favor TMZ entry into the cells, counteracting the drug extrusion through the membrane ABC protein, a mechanism through which TMZ loses its efficacy [74]. Moreover, since BzATP significantly reduced GSC accumulation in the G2/M phase of the cell cycle caused by TMZ, a period during which DNA repair may occur, a mitotic catastrophe might be favored, like that induced by resveratrol [75].
Of interest, the potentiated effect due to the combined administration of nucleotides plus TMZ to GSCs was long-lasting when TMZ was readministered. Likely, TMZ cytotoxicity was increased as it acted after a short period on cells damaged by the previous combined treatment with nucleotides and TMZ. This aspect, in our opinion, deserves further investigation, and we hypothesize that repeated treatments could be successful also in GSCs grown as neurospheres.
Thus, a lot of work remains to be done to assess the most appropriate doses and times of drug administration in order to optimize the antitumoral effect of purines either if administered alone or in combination with a classic antineoplastic agent. However, based on our results, we think that the investigation on purinoceptor ligands is promising and may open the way to further studies on in vivo tumor models.
References
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Clarke J, Butowski N, Chang S (2010) Recent advances in therapy for glioblastoma. Arch Neurol 67:279–283
Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, Reynolds BA (2011) Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 134(Pt 5):1331–1343
Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488(7412):522–526
Götz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6:777–788
Lois C, Alvarez-Buylla A (1993) Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A 90:2074–2077
Morshead CM, Craig CG, van der Kooy D (1998) In vivo clonal analyses reveal the properties of endogenous neural stem cell proliferation in the adult mammalian forebrain. Development 125:2251–2261
Berger F, Gay E, Pelletier L, Tropel P, Wion D (2004) Development of gliomas: potential role of asymmetrical cell division of neural stem cells. Lancet Oncol 5(8):511–514
Singh SK, Clarke ID, Hide T, Dirks PB (2004) Cancer stem cells in nervous system tumors. Oncogene 23:7267–7273
Pérez Castillo A, Aguilar-Morante D, Morales-García JA, Dorado J (2008) Cancer stem cells and brain tumors. Clin Transl Oncol 10:262–267
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Rathbone MP, Middlemiss PJ, Gysbers JW, Andrew C, Herman MAR, Reed JK, Ciccarelli R, Di Iorio P, Caciagli F (1999) Trophic effects of purines in neuron and glial cells. Prog Neurobiol 59:663–690
Burnstock G (2008) Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 7(7):575–590
Burnstock G, Ulrich H (2011) Purinergic signaling in embryonic and stem cell development. Cell Mol Life Sci 68(8):1369–1394
Zimmermann H (2006) Nucleotide signaling in nervous system development. Pflugers Arch 452:573–588
Rishton GM (2008) Small molecules that promote neurogenesis in vitro. Recent Patents CNS Drug Discov 3(3):200–208
Resende RR, Majumder P, Gomes KN, Britto LRG, Ulrich H (2007) P19 embryonal carcinoma cells as in vitro model for studying purinergic receptor expression and modulation of N-methyl-D-aspartate-glutamate and acetylcholine receptors during neuronal differentiation. Neuroscience 146:1169–1181
Migita H, Kominami K, Higashida M, Maruyama R, Tuchida N, McDonald F, Shimada F, Sakudara K (2008) Activation of adenosine A1 receptor-induced neural stem cells proliferation via MEK/ERK and Akt signaling pathway. J Neurosci Res 86:2820–2828
Jagasia R, Song H, Gage FH, Lie DC (2006) New regulators in adult neurogenesis and their potential role for repair. Trends Mol Med 12:400–405
Zang Y, Yu L-F, Pang T, Fang L-P, Feng X, Wen T-Q, Nan F-J, Feng L-Y, Li J (2008) AICAR induces astroglial differentiation of neural stem cells via activating the JAK/STAT3 pathway independently of AMP-activated protein kinase. J Biol Chem 283:6201–6208
White N, Burnstock G (2006) P2 receptors and cancer. Trends Pharmacol Sci 27:211–217
Robak T, Robak P (2012) Purine nucleoside analogs in the treatment of rarer chronic lymphoid leukemias. Curr Pharm Des 18(23):3373–3388
Miano M, Pistorio A, Putti MC, Dufour C, Messina C, Barisone E, Ziino O, Parasole R, Luciani M, Lo Nigro L, De Rossi G, Varotto S, Bertorello N, Petruzziello F, Calvillo M, Micalizzi C (2012) Clofarabine, cyclophosphamide and etoposide for the treatment of relapsed or resistant acute leukemia in pediatric patients. Leuk Lymphoma 53(9):1693–1698
Holowiecki J, Grosicki S, Giebel S, Robak T, Kyrcz-Krzemien S, Kuliczkowski K, Skotnicki AB, Hellmann A, Sulek K, Dmoszynska A, Kloczko J, Jedrzejczak WW, Zdziarska B, Warzocha K, Zawilska K, Komarnicki M, Kielbinski M, Piatkowska-Jakubas B, Wierzbowska A, Wach M, Haus O (2012) Cladribine, but not fludarabine, added to daunorubicin and cytarabine during induction prolongs survival of patients with acute myeloid leukemia: a multicenter, randomized phase III study. J Clin Oncol 30(20):2441–2448
Bellezza I, Tucci A, Minelli A (2008) 2-Chloroadenosine and human prostate cancer cells. Anticancer Agents Med Chem 8(7):783–789
Galmarini CM, Popowycz F, Joseph B (2008) Cytotoxic nucleoside analogues: different strategies to improve their clinical efficacy. Curr Med Chem 15(11):1072–1082
Kim H, Kang JW, Lee S, Choi WJ, Jeong LS, Yang Y, Hong JT, do Yoon Y (2010) A3 adenosine receptor antagonist, truncated thio-Cl-IB-MECA, induces apoptosis in T24 human bladder cancer cells. Anticancer Res 30(7):2823–2830
Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L, Fishman P (2008) The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappaB signal transduction pathways. Int J Oncol 33(2):287–295
Schafer R, Sedehizade F, Welte T, Reiser G (2003) ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am J Physiol Lung Cell Mol Physiol 285(2):L376–L385
Morrone FB, Jacques-Silva MC, Horn AP, Bernardi A, Schwartsmann G, Rodnight R, Lenz G (2003) Extracellular nucleotides and nucleosides induce proliferation and increase nucleoside transport in human glioma cell lines. J Neurooncol 64:211–218
Sellers LA, Simon J, Lundahl TS, Cousens DJ, Humphrey PP, Barnard EA (2001) Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem 276:16379–16390
Resende RR, Britto LRG, Ulrich H (2008) Pharmacological properties of purinergic receptors and their effects on proliferation and induction of neuronal differentiation of P19 carcinoma cells. Int J Dev Neurosci 26(7):763–777
Guarnieri S, Pilla R, Morabito C, Sacchetti S, Mancinelli R, Fanò G, Mariggiò MA (2009) Extracellular guanosine and GTP promote expression of differentiation markers and induce S-phase cell-cycle arrest in human SH-SY5Y neuroblastoma cells. Int J Dev Neurosci 27(2):135–147
Traversa U, Bombi G, Di Iorio P, Ciccarelli R, Werstiuk ES, Rathbone MP (2002) Specific [(3)H]-guanosine binding sites in rat brain membranes. Br J Pharmacol 135(4):969–976
Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H (2008) Purinergic signalling in the nervous system: an overview. Trends Neurosci 32:19–29
Burnstock G (2002) Potential therapeutic targets in the rapidly expanding field of purinergic signalling. Clin Med 2(1):45–53
Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors: an update. Pharmacol Rev 63(1):1–34
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58(3):281–341
Mishra SK, Braun N, Shukla V, Fullgrabe M, Schomerus C, Korf HW, Gachet C, Ikehara Y, Sevigny J, Robson SC, Zimmermann H (2006) Extracellular nucleotide signaling in adult neural stem cells: synergism with growth factor-mediated cellular proliferation. Development 133:675–684
Ledur PF, Villodre ES, Paulus R, Cruz LA, Flores DG, Lenz G (2012) Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal 8(1):39–48
Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA (2008) Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res 6:21–30
Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA (2006) Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9:391–403
Braganhol E, Wink MR, Lenz G, Battastini AM (2013) Purinergic signaling in glioma progression. Adv Exp Med Biol 986:81–102
Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, Pilozzi E, Larocca LM, Peschle C, De Maria R (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13(7):1238–1241
Pallini R, Ricci-Vitiani L, Banna GL, Signore M, Lombardi D, Todaro M, Stassi G, Martini M, Maira G, Larocca LM, De Maria R (2008) Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res 14(24):8205–8212
Griffero F, Daga A, Marubbi D, Capra MC, Melotti A, Pattarozzi A, Gatti M, Bajetto A, Barbieri F, Favoni RE, Lo Casto M, Zona G, Spaziante R, Florio T, Corte G (2009) Different response of human glioma tumor-initiating cells to Epidermal Growth Factor receptor kinase inhibitors. J Biol Chem 284(11):7138–7148
Angelucci S, Marchisio M, Di Giuseppe F, Pierdomenico L, Sulpizio M, Eleuterio E, Lanuti P, Sabatino G, Miscia S, Di Ilio C (2010) Proteome analysis of human Wharton’s jelly cells during in vitro expansion. Proteome Sci 8:18
Vindeirinho J, Costa GN, Correia MB, Cavadas C, Santos PF (2013) Effect of diabetes/hyperglycemia on the rat retinal adenosinergic system. PLoS One 8(6), e67499
Sim JA, Young MT, Sung HY, North RA, Surprenant A (2004) Reanalysis of P2X7 receptor expression in rodent brain. J Neurosci 24(28):6307–6314
Alvarenga EC, Rodrigues R, Caricati-Neto A, Silva-Filho FC, Paredes-Gamero EJ, Ferreira AT (2010) Low-intensity pulsed ultrasound-dependent osteoblast proliferation occurs by via activation of the P2Y receptor: role of the P2Y1 receptor. Bone 46(2):355–362
Zhu CB, Lindler KM, Campbell NG, Sutcliffe JS, Hewlett WA, Blakely RD (2011) Colocalization and regulated physical association of presynaptic serotonin transporters with A3 adenosine receptors. Mol Pharmacol 80(3):458–465
Montano N, Cenci T, Martini M, D’Alessandris QG, Pelacchi F, Ricci-Vitiani L, Maira G, de Maria R, Larocca LM, Pallini R (2011) Expresion of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia 13(12):1113–1121
Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, Sawaya R, Aldape K (2005) Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 11:1462–1466
Hegi ME, Diserens AC, Gorlia T, Hamou MF, deTribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352:997–1003
Roger S, Pelegrin P (2011) P2X7 receptor antagonism in the treatment of cancers. Expert Opin Investig Drugs 20(7):875–880
Souza CO, Santoro GF, Figliuolo VR, Nanini HF, de Souza HS, Castelo-Branco MT, Abalo AA, Paiva MM, Coutinho CM, Coutinho-Silva R (2012) Extracellular ATP induces cell death in human intestinal epithelial cells. Biochim Biophys Acta 1820(12):1867–1878
Vlachostergios PJ, Hatzidaki E, Papandreou CN (2013) MGMT repletion after treatment of glioblastoma cells with temozolomide and O6-benzylguanine implicates NFκB and mutant p53. Neurol Res 35(8):879–882
Ceruti S, Abbracchio MP (2013) Adenosine signaling in glioma cells. Adv Exp Med Biol 986:13–30
Tafani M, Di Vito M, Frati A, Pellegrini L, De Santis E, Sette G, Eramo A, Sale P, Mari E, Santoro A, Raco A, Salvati M, De Maria R, Russo MA (2011) Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J Neuroinflammation 8:32
Madi LOA, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P (2004) The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin Cancer Res 10(13):4472–4479
Anccasi RM, Ornelas IM, Cossenza M, Persechini PM, Ventura AL (2013) ATP induces the death of developing avian retinal neurons in culture via activation of P2X7 and glutamate receptors. Purinergic Signal 9(1):15–29
Kim TH, Kim YK, Woo JS (2012) The adenosine A3 receptor agonist Cl-IB-MECA induces cell death through Ca(2+)/ROS-dependent down regulation of ERK and Akt in A172 human glioma cells. Neurochem Res 37(12):2667–2677
Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA (2012) Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov Today 17(7-8):359–366
Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, Bianchi G, Kroemer G, Pistoia V, Di Virgilio F (2012) Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res 72(12):2957–2969
Glaser T, Cappellari AR, Pillat MM, Iser IC, Wink MR, Battastini AM, Ulrich H (2012) Perspectives of purinergic signaling in stem cell differentiation and tissue regeneration. Purinergic Signal 8(3):523–537
Taliani S, La Motta C, Mugnaini L, Simorini F, Salerno S, Marini AM, Da Settimo F, Cosconati S, Cosimelli B, Greco G, Limongelli V, Marinelli L, Novellino E, Ciampi O, Daniele S, Trincavelli ML, Martini C (2010) Novel N2-substituted pyrazolo[3,4-d]pyrimidine adenosine A3 receptor antagonists: inhibition of A3-mediated human glioblastoma cell proliferation. J Med Chem 53(10):3954–3963
Daniele S, Zappelli E, Natali L, Martini C, Trincavelli ML (2014) Modulation of A1 and A2B adenosine receptor activity: a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell Death Dis 5, e1539
Leij-Halfwerk S, Agteresck KJ, Sijens PE, Dagnelie PC (2002) Adenosine triphosphate infusion increases liver energy status in advanced lung cancer patients: an in vivo 31P magnetic resonance spectroscopy study. Hepatology 35(2):421–424
Beijer S, van Rossum E, Hupperets PS, Spreeuwenberg C, van den Beuken M, Winkens RA, van den Borne BE, de Graeff A, Dagnelie PC (2007) Application of adenosine 5’-triphosphate (ATP) infusions in palliative home care: design of a randomized clinical trial. BMC Public Health 7:4
Shabbir M, Burnstock G (2009) Purinergic receptor-mediated effects of adenosine 5’-triphosphate in urological malignant diseases. Int J Urol 16:143–150
Barberà-Cremades M, Baroja-Mazo A, Gomez AI, Machado F, Di Virgilio F, Pelegrin P (2012) P2X7 receptor-stimulation causes fever via PGE2 and IL-1β release. FASEB J 26(7):2951–2962
Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M (2009) Systemic administration of an antagonist of the ATP-senstive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A 106(30):12489–12493
Wei Q, Costanzi S, Liu QZ, Gao ZG, Jacobson KA (2011) Activation of the P2Y1 receptor induces apoptosis and inhibits proliferation of prostate cancer cells. Biochem Pharmacol 82(4):418–425
Peigñan L, Garrido W, Segura R, Melo R, Rojas D, Cárcamo JG, San Martín R, Quezada C (2011) Combined use of anticancer drugs and an inhibitor of multiple drug resistance-associated protein-1 increases sensitivity and decreases survival of glioblastoma multiforme cells in vitro. Neurochem Res 36(8):1397–1406
Filippi-Chiela EC, Thome MP, Bueno e Silva MM, Pelegrini AL, Ledur PF, Garicochea B, Zamin LL, Lenz G (2013) Resveratrol abrogates the temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the temozolomide-induced senescence in glioma cells. BMC Cancer 13:147
Acknowledgments
The authors declare no conflict of interest. Study supported by a grant to R.C. from the Italian Government (PRIN, prot. 2008H42W5X_002) and in part by a grant to StemTeCh Group from CARICHIETI Foundation. We thank Prof. Marco Marchisio and Dr. Laura Pierdomenico (Dept. of Medicine and Aging Sciences, University of Chieti) for their invaluable support for the experiments in which cytofluorimetric analysis was used to determine the cell cycle distribution.
Author information
Authors and Affiliations
Corresponding author
Additional information
Francesco Caciagli and Renata Ciccarelli contributed equally to this work.
Rights and permissions
About this article

Cite this article
D’Alimonte, I., Nargi, E., Zuccarini, M. et al. Potentiation of temozolomide antitumor effect by purine receptor ligands able to restrain the in vitro growth of human glioblastoma stem cells. Purinergic Signalling 11, 331–346 (2015). https://doi.org/10.1007/s11302-015-9454-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-015-9454-7