
Abstract
Astrocytes are fundamental for central nervous system (CNS) physiology and are the fulcrum of neurological diseases. Astroglial cells control development of the nervous system, regulate synaptogenesis, maturation, maintenance and plasticity of synapses and are central for nervous system homeostasis. Astroglial reactions determine progression and outcome of many neuropathologies and are critical for regeneration and remodelling of neural circuits following trauma, stroke, ischaemia or neurodegenerative disorders. They secrete multiple neurotransmitters and neurohormones to communicate with neurones, microglia and the vascular walls of capillaries. Signalling through release of ATP is the most widespread mean of communication between astrocytes and other types of neural cells. ATP serves as a fast excitatory neurotransmitter and has pronounced long-term (trophic) roles in cell proliferation, growth, and development. During pathology, ATP is released from damaged cells and acts both as a cytotoxic factor and a proinflammatory mediator, being a universal “danger” signal. In this review, we summarise contemporary knowledge on the role of purinergic receptors (P2Rs) in a variety of diseases in relation to changes of astrocytic functions and nucleotide signalling. We have focussed on the role of the ionotropic P2X and metabotropic P2YRs working alone or in concert to modify the release of neurotransmitters, to activate signalling cascades and to change the expression levels of ion channels and protein kinases. All these effects are of great importance for the initiation, progression and maintenance of astrogliosis–the conserved and ubiquitous glial defensive reaction to CNS pathologies. We highlighted specific aspects of reactive astrogliosis, especially with respect to the involvement of the P2X7 and P2Y1R subtypes. Reactive astrogliosis exerts both beneficial and detrimental effects in a context-specific manner determined by distinct molecular signalling cascades. Understanding the role of purinergic signalling in astrocytes is critical to identifying new therapeutic principles to treat acute and chronic neurological diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuroglia of the central nervous system (CNS) is represented by highly heterogeneous cellular populations generally classified as astrocytes, oligodendrocytes, microglia, and chondroitin sulphate proteoglycan NG2-positive glia (also known as synantocytes; [1]). Adult brain also contains radial Müller cells in the retina and pseudoradial Bergmann glial cells in the cerebellum [2, 3]. Astrocytes, oligodendrocytes and NG2-glial cells are of ectodermal/neural origin, whereas microglial cells of mesodermal origin invade the CNS during perinatal period. Neuroglial cells perform multiple functions being generally responsible for homeostasis of the nervous system at multiple levels including neurogenesis and development of the CNS, defining nervous system cytoarchitecture, providing metabolic support, controlling molecular homeostasis of the extracellular space and mounting CNS defence. Extracellular adenosine 5′-triphosphate (ATP) plays a complex signalling role in the CNS, and its binding to P2 purinoceptors (P2Rs) can be neuroprotective or neurotoxic in various pathological conditions. In this review, we shall summarise the role of nucleotide signalling in astroglia under physiological and pathophysiological conditions.
The ubiquity of the extracellular signalling molecule ATP and diversity of purinoceptors allow multiple physiological roles (including development and growth) and pathological significance of the purinergic signalling system [4–12]. ATP, released from neurones and glial cells, contributes to synaptic transmission in many brain regions. Physiologically ATP is delivered by exocytosis of ATP-containing vesicles or vesicles co-storing ATP with other neurotransmitters such as glutamate, γ-aminobutyric acid (GABA), noradrenaline or acetylcholine (ACh) [9, 13–17]. Regulated release of ATP is relevant for neuronal–glial communications (for example regulating synaptic plasticity) as well as for glial–glial signalling (for example, by initiating propagating Ca2+ waves) [9, 18].
Pathological insults to the CNS, e.g., hypoxia, ischaemia, trauma or epilepsy-associated seizures are accompanied by the rapid increase in concentrations of extracellular purine nucleotides resulting from massive exit of ATP from damaged neural cells; in addition, purines can be released by exocytotic and transporter-mediated processes [19–23]. Increases in extracellular purines may assume cytotoxic proportions resulting in cell death.
The released ATP undergoes rapid and successive enzymatic degradation by ectonucleotidases, producing adenosine diphosphate (ADP), adenosine monophosphate (AMP) and adenosine. Adenosine also acts as a signalling molecule of its own right in the CNS and often has effects opposite to those of ATP [9, 24, 25].
Purine and pyrimidine nucleotides/nucleosides activate several families of purinoceptors, classified as metabotropic P1 adenosine receptors, ionotropic P2XRs and metabotropic P2YRs, which are broadly distributed in both neurones and glial cells; activation of these receptors mediate a remarkable variety of physiological and pathophysiological reactions [5, 11, 26–31]. Ionotropic (cationic) P2XRs are represented by seven subtypes (P2X1 through P2X7). Importantly, permeability of P2X2, P2X4, and P2X7Rs to larger cations increases following repetitive or longer lasting exposure to ATP and (particularly in the case of P2X7Rs) may open large pores in the cell membrane. There are eight metabotropic G-protein-coupled P2YRs, with different ligand preferences (P2Y1: ADP; P2Y2: ATP/UTP; P2Y4: UTP; P2Y6: UDP; P2Y11: ATP; P2Y12: ADP; P2Y13: ADP and P2Y14: UDP-glucose and other nucleotide sugars).
Astrocytes in the normal brain
Functions of astrocytes
Astrocytes are fundamental for normal brain physiology and, being homeostatic/defensive cellular elements of the brain, are deeply involved in neuropathology, to a very great extent determining the progress and outcome of various neurological diseases [9, 32–37].
Astrocytes have a highly heterogeneous morphological appearance. Main types of astrocytes are represented by (1) protoplasmic astrocytes of grey matter, which possess numerous highly branched fine processes, and (2) the fibrous astrocytes in the white matter, which have less branched and thicker processes. In the cortex, astrocytes occupy strictly delineated domains, within astrocytes form multiple contacts with neuronal structures, whereas astroglial perivascular end-feet envelop CNS blood vessels.
In the grey matter, astroglial perisynaptic processes ensheath synapses, whereas in the white matter astroglial processes terminate at nodes of Ranvier, the sites of action potential generation [37]. Astrocytic processes associated with synapses tightly regulate synapse formation, maintenance and plasticity [38, 39]. For example, in the hippocampus or cortex of rodents, finely branching processes emanating from a single astrocyte are estimated to contact several hundred dendrites from multiple neurones and to envelope 20,000–100,000 synapses [36]. Interest in astroglia function has increased steadily in recent years because of their newly discovered roles in synapse formation, maturation, efficacy and plasticity [40].
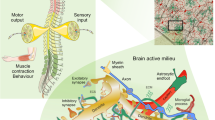
Astrocytes express intermediate filament proteins [e.g. glial fibrillary acidic protein (GFAP), vimentin, nestin] depending on their type and developmental stage. There are different isoforms and splice variants of GFAP, including GFAP α, β, γ, δ, and κ, which are expressed in a heterogeneous manner in healthy CNS and in pathological tissues including gliomas [36]. GFAP and vimentin, together with microtubules and actin filaments, constitute the cytoskeleton of astrocytes. Expression of GFAP is often up-regulated in pathophysiological stages; this up-regulation is generally considered as hallmark of reactive astrogliosis, the latter being evolutionary conserved and ubiquitous defensive reaction of astroglia [36, 41]. Some examples of astroglial activation after mechanical injury in vitro and in vivo are given in Fig. 1.

From astroglial cell cultures to the human brain—up-regulation of P2Y1R expression on activated GFAP-astrocytes. Confocal images of double immunofluorescence to characterise the expression of P2Y1R subtypes on activated astrocytes after mechanical injury in a–f in vitro, g–i in vivo in rat brain and j–l in vivo in human brain. Under physiological conditions (data not shown) in rat and human tissue the expression of P2Y1R on single astrocytes was observed, but with low intensity. In vitro (primary cultures of rat cortical astrocytes): a, d overview about GFAP-positive astrocytes after mechanical injury (marked by asterisks in a; a near the lesion, d far away from the lesion). b, c Single outgrowing astrocytes in the lesioned area with clear co-expression of the astrocytic marker GFAP with the P2Y1R subtype (arrows); d a high number of GFAP/P2Y1R-positive astrocytes at sites remote from the lesion; e, f one example of the co-localisation in higher magnification. In vivo (rat brain, cortex, perilesion area): g overview about the perilesion area after mechanical injury (stab wound injury; according [100]; marked by asterisks) in rat cortex. h, i Single activated GFAP-positive astrocytes in higher magnification with clear co-expression with the P2Y1R subtype (arrows). In vivo (human brain; removed at autopsy, pericontusion zone, cortex): j–l After traumatic brain injury (j, overview, pericontusion zone), the P2Y1R was found on activated GFAP-positive astrocytes (k, l; arrows) in the pericontusion zone around the injured area (K. Bremicker, M. Weber and H. Franke, unpublished data)
Astrocytes assume numerous and well-defined supportive functions, including structural support, providing for neurovascular coupling, regulation of extracellular K+, uptake of neurotransmitters, metabolic support of neurones, etc. Astroglia integrate glia, neurones and brain vasculature into functional networks. Pathological potential of astrocytes is similarly manifold, including the formation of neuroprotective scars following injury, homeostasis preservation, blood–brain barrier maintenance and repair, axon guidance during regeneration as well as responses to neuronal activity and plasticity. Astrocytes are able to sense and modulate neuronal environment and to actively remodel themselves to meet constantly changing neuronal demands (e.g. [16, 42, 43]).
Astrocytes are also central for energy consumption, neurotransmitter release and reactive oxygen species (ROS) production in the nervous tissue. Astroglia regulate (1) the uptake of glucose and the release of lactate (“astrocyte-neurone-lactate shuttle”), (2) the uptake of glutamate and the release of glutamine (“glutamate-glutamine cycle” maintaining both glutamatergic and GABAergic synaptic transmissions) and (3) the uptake of glutathione precursors and the release of glutathione [42–45]. Glutamate, released into the synaptic cleft during neuronal activity, is rapidly removed by surrounding astrocytes, which convert glutamate to glutamine (using the enzyme glutamine synthase) and release glutamine into the interstitial space for uptake by neurones [46]. The uptake of synaptic glutamate by astroglia is the major mechanism preventing accumulation of glutamate in the synaptic space and thus protecting neurones from excessive activation and excitotoxic cell death [42]. At the same time, glutamate–glutamine shuttle is the only pathway allowing neurones to replenish their stores of glutamate and GABA.
Nucleotide-induced modulation of astrocytic function
Astrocytes are arguably the main source for the physiologically released ATP in the CNS [47]. They release ATP to communicate with each other and with other types of glia by stimulating purinoceptors [9, 10, 17, 18, 48]. Astroglial ATP and its metabolite adenosine activate purinoceptors on neurones, microglia, oligodendrocytes and blood vessels [37]. Generally, ATP and other nucleotides are taken up by and stored in secretory and synaptic vesicles. Accumulation of ATP into neuronal synaptic vesicles is mediated by a Cl−-dependent vesicular nucleotide transporter expressed in astroglia [49, 50]. Conceptually, ATP can be released from astrocytes in several ways by (1) Ca2+-dependent exocytosis [14, 15, 51, 52], (2) from lysosomes [53], and (3) through hemichannels formed by connexins and pannexins (note activation by ATP of pannexin-1 channels and involvement of P2YRs [54]; P2X7Rs [55], as well as connexin-43 channels and involvement of P2YRs [56, 57]). In addition, plasmalemmal voltage-dependent anion channels and P2X7Rs can function as exit pathways for glutamate or ATP release [9, 14, 37, 58].
Astrocytes communicate with each other via intercellular “calcium waves” [9, 16, 18, 59–63]. These cells respond to a wide range of physiological and pathophysiological stimuli with increases in cytosolic Ca2+ ([Ca2+]i) mainly mediated through Gq protein-coupled receptors, including those of the P2Y type, with subsequent release of Ca2+ from intracellular stores through inositol 1,4,5-trisphosphate (InsP3) receptors. Propagation of Ca2+ waves results from the intercellular diffusion of InsP3 through gap junctions or from the release of ATP in a single cell, this release then producing [Ca2+]i transients in the neighbouring cell by activation of P2YR/InsP3 signalling systems [47, 64, 65].
Astrocytes also release several non-nucleotide neurotransmitters, including adenosine, glutamate and d-serine, to stimulate their respective receptors at neighbouring neurones and glial cells [66–69]. Ca2+ waves underlie communication in astrocytic networks, being the substrate of glial excitability.
In the CNS, the synapses are generally organised according to “tripartite” architecture, according to which the synapse is composed of the presynaptic neuronal terminal, the postsynaptic membranes of dendrites or neuronal somata, and of the astrocytic perisynaptic process enwrapping neuronal structures. Within the “tripartite” synapse, ATP released during synaptic transmission activates astrocytic receptors, which initiate Ca2+ signals and Ca2+ waves in the astroglial networks. Astroglial Ca2+ signals in turn induce the release of ATP, which may feed back to neurones via activation of pre-, postsynaptic and extrasynaptic P1 and P2Rs [9, 70–74].
P2 receptors at astrocytes
Astrocytic P2X and P2Y receptors
Astrocytes express various types of functional P2X/Y purinoceptors (e.g. [4, 5, 37, 75–87]). Nucleotide signalling mediated by these receptors is of significance for both physiological regulation and pathophysiological processes in the nervous system [6, 88, 89]. Ionotropic P2XRs are responsible for rapid astrocytic signalling, whereas G-protein-coupled P2YRs mediate long-term effects, including trophic responses through a variety of intracellular pathways including gene activation [7, 12, 90].
Glial P2X7Rs are of special importance, since these receptors are involved in initiation of apoptosis [91–93], and act as “emergency” receptors aimed at restraining the excessive astrogliosis triggered by brain injuries [94, 95]. It is noteworthy that ionotropic P2X7Rs, through their ability to open large pores in the cell membranes, are able to damage the cytoskeleton; they appear also to trigger necrotic/apoptotic death of neurones and glia by regulating the processing and release of interleukin-1β (IL-1β), a key mediator in neurodegeneration, chronic inflammation and chronic pain [96, 97].
Recently P2X7R-mediated currents were identified in cultured rodent cortical astrocytes [58, 85] and in astrocytes patch-clamped in acute brain slices of rats and mice [86]. The discovery of functional P2X7Rs in astrocytes of the prefrontal cortex and hippocampus is somewhat at odds to the previously reported failure of ATP and BzATP to activate ion currents in astrocytes in hippocampal slices of transgenic mice that express enhanced green fluorescent protein (EGFP) under the control of the human glial fibrillary acidic promoter [Tg(GFAP/EGFP)] [98]. There are at least two possible reasons for this discrepancy. First, in contrast to the prefrontal cortex, the BzATP-induced astrocytic currents in the hippocampus are minuscule, when measured in a normal Ca2+/Mg2+-containing external medium [86], although they can be greatly increased by exchanging the normal medium to one which contains a low Ca2+concentration with no added Mg2+. Second, the BzATP responses are also surprisingly very small in cortical astrocytes of Tg(GFAP/EGFP) mice and might have therefore interfered with their identification in the hippocampus. These results are in agreement with the reported data that both BzATP and ATP activate mouse recombinant P2X7R with much lower potencies than the corresponding rat receptor [99].
The presence of multiple P2X and P2Y receptors at the same astrocyte may lead to an abundance of effects, the common denominator arguably being an increase in [Ca2+]I, which may trigger further second messenger pathways resulting in the neurodegenerative or proliferative reactions [80, 100, 101].
Interaction of astrocytic P2 receptors with other receptors
P2YRs may assemble as homodimers or heterodimers with other P2YRs or with other G protein-coupled transmitter receptors in glia and neurones [31, 102]. For example, adenosine A1 and nucleotide P2Y1Rs aggregate to form a multimeric complex in human astrocytes and the regulation of A1R function is induced by P2Y1R activation [103]. A high level of co-localisation between A1Rs and P2Y1Rs at glutamatergic synapses and surrounding astrocytes and the functional interaction between these receptors in the hippocampus have been demonstrated [104]. P2Y1R stimulation impaired the potency of A1R coupling to G proteins, whereas the stimulation of A1Rs increased the functional responsiveness of P2Y1Rs. The interaction between A1 and P2Y1Rs is important for nucleotide signalling in astrocytes because it is involved in cell-to-cell communication and in the control of synaptic transmission, particularly during pathological conditions, when large amounts of purines are released [103]. In hippocampal astrocytes, P2Y1 and P2Y2R-mediated Ca2+ responses exhibit two forms of activity-dependent negative feedback mechanisms on synaptic transmission via the phospholipase (PL)Cβ-InsP3 pathway [79].
P2YRs also interact with the ionotropic P2XRs either directly or indirectly. P2Y2 and P2X7R stimulation of protein kinase D (PKD) activation and localisation in primary rat cerebellar astrocytes was demonstrated [105], and the contribution of calcium-permeable heteromeric P2X1/5R channels to the excitability of astrocytes appeared to be modulated by PI(4,5)P(2) through the P2X1 lipid-binding domain [106].
Astrocytes in the injured CNS
Reactive gliosis and purinergic modulation
Astrogliosis is fundamental for regeneration following lesioning of the brain. In vitro and in vivo experiments corroborated the important role of nucleotides in the reactive responses of astrocytes to brain injury because purinoceptors regulate morphological remodelling, proliferation, up-regulation of GFAP synthesis as well as chemotaxis and chemokinesis in activated astrocytes.
Different kinds of astrogliosis have been described [44], which are classified as follows: (1) anisomorphic astrogliosis–often observed as an encapsulation of the damaged CNS region after neurotrauma; it is characterised by disappearance of astroglial domain organisation and the formation of a dense plexus or wall; (2) isomorphic astrogliosis–characterised by astroglial hypertrophy with preserved domain organisation and is often reversible and beneficial for postraumatic regeneration.
Various glial cells and cellular signals are involved in the formation of the glial scar representing the end stage of anisomorphic gliosis. Within the first few hours after injury, neuronal and glial cells undergo cell death at the injury site, with a concurrent recruitment of CNS microglia and peripheral monocytes (including macrophages [107]). Astrocytes migrate from the adjacent undamaged parenchyma towards the injured cells and start the reparation, for example by protruding processes into the injury core [108–111].
Immediately after ischaemic or traumatic damage to the brain, extracellular ATP concentrations markedly increase not only because of lack of efficient metabolism by degrading enzymes but also due to active release from surviving cells [21, 23, 112]. One of the major hallmarks of nucleotide-induced reactive astrogliosis is that astrocytes react to ATP with hypertrophy, swelling of the cell body and main processes, and proliferation that ultimately result in full blown astrogliotic phenotype with subsequent formation of the glial scar [88, 113, 114]. Astrocytes react to nucleotides with up-regulation of the GFAP and vimentin. In cultured astrocytes, it was repeatedly shown that extracellular ATP and its structural analogues increase GFAP content and DNA synthesis [115, 116]. Morphological remodelling (e.g. “stellation” or an increase in the mean length of processes of GFAP positive cells; see also examples given in Fig. 1A, B) was also described in vitro [115, 117–119].
Within 3–5 days after injury, hypertrophic and elongated astrocytes at the injury border express high levels of vimentin and brain lipid-binding protein (reviewed in [35]). The mitogenic and morphogenic responses to extracellular ATP in cultured astrocytes have been well documented [88, 101, 113, 120–124]. Proliferation of reactive astrocytes, as indicated by bromodeoxyuridine (BrdU) incorporation or the expression of Ki67, is stimulated by ATP and P2R agonists in vivo and in vitro (e.g. [125–128]). The total number of astrocytes (identified e.g. by GFAP or S100β labelling) increases over time and in correlation with the expression of additional nucleotide receptors (e.g. [23, 129]).
Although it has been shown that nestin-positive progenitor cells differentiate into astrocytes at the site of injury [44], the extent to which these cells contribute to the formation of the astroglial scar remains open. Similarly, we do not know the intrinsic and extrinsic mechanisms by which mature astrocytes contribute to the formation of precursor cells in the process of reactive gliosis [35].
Mediators of reactive gliosis
Astrocytes exhibit both regional and local variability that is dynamically regulated by a large number of inter- and intracellular signalling molecules. Acute brain injury is a complex phenomenon and ATP is only one of many endogenous factors determining reactive astrogliosis (acting alone or in concert with other factors).
There are many potential intercellular signalling molecules that are able to trigger reactive astrogliosis or to regulate specific aspects of it; these molecules are large polypeptide growth factors and cytokines [such as interleukins (IL-1, IL-6, and IL-10), ciliary neurotrotrophic factor (CNTF), tumour necrosis factor-α (TNF-α), interferon-γ (INFγ), transforming growth factor-β (TGFβ), fibroblast growth factor-2 (FGF2), leukaemia inhibitory factor (LIF), etc.], mediators of innate immunity (e.g. lipopolysaccharide [LPS], other Toll-like receptor ligands), neurotransmitters (e.g. ATP, glutamate, noradrenaline), ROS, nitric oxide (NO), factors released in consequence of hypoxia and glucose deprivation [e.g. monocyte chemotactic protein-1/chemokine ligand 2 (MCP-1/CCL2) and interferon-inducible protein-10/chemokine ligand 10 (IP-10/CXCL10)], products associated with neurodegeneration (e.g. β-amyloid) and eventually molecules associated with systemic metabolic toxicity or regulators of cell proliferation (e.g. endothelin-1) [36, 44, 101, 130–132]. Purines interact with these factors in complex manners.
It has been suggested that under pathological conditions, such as chronic inflammation and neurodegeneration, astroglia switch their phenotypes from metabolically supportive cells to immunocompetent cells capable of inducing inflammation via the production of a variety of proinflammatory factors [44]. ATP release during neuronal excitation or injury can enhance the inflammatory effects of cytokines (IL-1β, TNF-α, and IFN-γ) and prostaglandin E2 (PGE2) in cultures of astrocytes and may contribute to chronic inflammation [133, 134].
It has been demonstrated that the selective stimulation of astroglial P2Y1Rs is involved in the secretion of glutamate (e.g. [68, 135]) and the regulation of several cytokines/chemokines (for example, IL-6 [136], glial cell line-derived neurotrophic factors (GDNF) [137], IL-6, TNF-α, monocyte chemotactic protein-1/chemokine (MCP-1/CCL2) and IP-10/CXCL10 messenger RNA (mRNA) expression [132]).
ATP can act in combination with fibroblast growth factor (FGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF) as well as nerve growth factor (NGF) to stimulate astrocytic proliferation, contributing to the process of reactive astrogliosis and to hypertrophic/hyperplastic responses [138]. ATP released from damaged cells and acting via P2YRs synergistically enhances the proliferative effects of FGF2, whereas P2XRs inhibit the ability of FGF2 to stimulate DNA synthesis in rat cortical astrocyte cultures [139]. Opposing effects of P2X and P2YRs on the ability of FGF2 to induce proliferation in primary cultures of rat cortical astrocytes were found. Low concentrations of ATP enhanced DNA synthesis induced by FGF2, whereas high concentrations inhibited FGF2-induced proliferation [140]. It was concluded that P2YRs enhance proliferation caused by FGF2 in astrocytes, whereas stimulation of P2X7Rs inhibits proliferation by shifting cells to a state of reversible growth arrest that may be mediated by protein kinase signalling [140].
Trauma-induced activation of P2Y4Rs in astrocytes stimulates synthesis and release of thrombospondin-1, an extracellular matrix molecule that induces synapse formation during development and might have a role in CNS repair and remodelling after injury [141]. ATP stimulates N-cadherin expression, which is a Ca2+-dependent cell-adhesion molecule involved in glia–glia and axon–glia interactions [142]. Chondroitin sulphate proteoglycans [143], tenascin-R [144], keratin, vimentin [145] and Eph receptor tyrosine kinase [146] are among the repellent proteins expressed by reactive astrocytes.
Functional consequences of reactive astrogliosis
The glial scar demarcates the healthy brain from the wounded area. In fact, recent experimental evidence indicates that astroglial scars act as neuroprotective barriers to inflammatory cells and infectious agents and that they are formed in particular along borders to severe tissue damage, necrosis, and infection or autoimmune-triggered inflammatory infiltration [36]. However, such a demarcation has also the consequence that reparative processes such as axonal outgrowth are inhibited, and therefore, either beneficial or detrimental outcomes might be favoured [113, 114].
Conceptually the glial scar protects CNS tissue by (1) uptake of potentially excitotoxic glutamate, (2) by counteracting oxidative stress via glutathione production, (3) by the release of adenosine, (4) by the degradation of amyloid-β (Aβ) peptides, (5) by the facilitation of blood brain barrier repair, (6) by the reduction of vasogenic oedema after trauma, stroke or obstructive hydrocephalus, (7) by the stabilisation of extracellular fluid and ion balance and (7) by the limitation of the spread of inflammatory cells or infectious agents from the areas of damage or disease into healthy CNS parenchyma (e.g. [36, 43, 45, 130]). At the same time, in spite of these multiple protective functions, reactive astrogliosis and scar formation might delay or inhibit regenerative responses; therefore, this astrocytic reaction may play an important role in the pathogenesis and progression of diverse neuropathological conditions.
In chronic neuropathologies, such as for example Alzheimer’s disease (AD), reactive astrocytes contribute and reinforce inflammatory cascades [45]. The inflammatory response occurs mainly around the amyloid plaques and is characterised by the release of proinflammatory substances from activated microglia and astroglia. The evolving general concept assumes that in the early stages of injury reactive astrocytes are needed to limit tissue damage including scar formation, which provides beneficial effects in shielding the brain, whereas the consequences of persistent reactive astrogliosis can be harmful.
ATP released by astrocytes mediates activation of microglia [147]. The functions of reactive astrocytes are partially overlapping with those of reactive microglia, the latter being specialised in phagocytosis and integration of inflammatory responses [35]. Microglial activation is essential for phagocytosis of cellular debris following injury and is important for the immunoprotection of the CNS [148]. ATP may act as a chemoattractant for microglia, directing them to a site of injury [96]. ATP released from activated astrocytes stimulates microglia as well, and microglial cells encounter high levels of ATP near dying and disintegrating cells. Collectively, these observations indicate that ATP acts on microglia through P2X7R, which serve as an important component of a neuroinflammatory response. Additionally, astroglial death induced by ATP-mediated microglial activation may be an important pathophysiological pathway in epileptogenesis [149]. It has been speculated that antagonists of the P2X7R could thus have therapeutic relevance in the treatment of AD, cerebral ischaemia and neuroinflammatory conditions by regulating pathologically activated glial cells [96].
Finally, microglia express multiple P2X/YRs, of which the P2X4, P2Y6 and P2Y12R subtypes play important roles in the pathophysiology of microglial cells after acute injury or neurodegenerative disorders, contributing to the nucleotide responses [e.g. 31, 150, 151].
Astroglia and microglia may also exert opposite effects. Recent findings indicate that microglia control astroglial proliferation by preventing the proliferative response to ATP/ADP and favouring an inhibitory effect of UTP/UDP. Several microglial P2YRs may be involved by inducing the release of messengers that restrain astrogliosis [152].
P2 nucleotide receptors and signalling cascades
Intracellular signalling cascades: the P2X (P2X7) receptor
Astroglial P2XRs can be activated by synaptic transmission and can mediate fast local signalling through the elevation of cytoplasmic Ca2+ and Na+ concentrations. A number of functional roles of astroglial P2X7Rs might be related to Ca2+ signalling (e.g. [91–93, 153]) and secretory activities of astrocytes [46, 154], which are promoting the further release of purines [91] via the receptor channel itself or an accessory protein. ATP-stimulated glutamate [46, 58] and GABA release [155] may also occur [96]. These signals can be translated into various physiological messages by numerous pathways, including release of neurotransmitters, metabolic support of neurones and regulation of activity of postsynaptic glutamate and GABAARs [10, 17]. P2XRs can interact with cell-adhesion molecules, physically cross-talk with other receptor channels such as nicotinic acetylcholine, GABAA or 5-HT3 receptor-channels, or interact with G protein-coupled receptors. Activation of P2XRs can also trigger intracellular trophic signalling pathways [12].
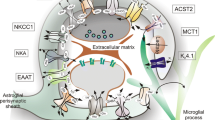
The P2X7R activates a particularly wide range of signal cues (Table 1 and Fig. 2). By virtue of its extended C terminus, it undergoes multiple protein interactions forming a signalling complex. P2X7Rs activate several second messenger and enzyme cascades, including extracellular signal-regulated protein kinases (ERKs), serine-thereonine kinase Akt (Akt), c-Jun N terminal kinases (JNKs) and p38 kinase. The stimulation of this receptor also increases protein tyrosine phosphorylation, ultimately leading to mitogen-activated protein kinase (MAPK) pathway initiation. In addition, by dilation of the channel pore, P2X7Rs can induce a collapse of ion gradients over the plasma membrane leading to cell death (see above [96, 156, 157]).

Schematic illustration of examples of signal transduction pathways in astroglial cells following P2X7R activation. After channel opening the P2X7R is permeable for Na+, K+ and Ca2+. Activation of the P2X7R triggers the efflux of K+ from cells and activates IL-1 converting enzyme, leading to cleavage of pro-IL-1β to mature IL-1β and release from the cell. Many events downstream of P2X7R activation are dependent on extracellular calcium influx. Stimulation of ionotropic P2X7Rs leads to activation of phospholipases A2 and D (PLA2, D) and protein kinase C (PKC), e.g. resulting in the activation of glycogen synthase kinase 3 (GSK3) or the activation of caspase cascades. Furthermore, the induction of second messenger and enzyme cascades promoted e.g. the activation of mitogen activated protein kinase (MAPK) pathway proteins (ERK1/2), p38 MAPK, and c-Jun N-terminal kinase (JNK) as well as PI3K/Akt activation. The activity of transcription factors, such as nuclear factor κB (NF-κB), cyclic element-binding protein (CREB), and activator protein (AP-1) are also up-regulated, leading to the expression of proinflammatory genes, such as cyclooxygenase-2 (COX-2) or inducible nitric oxide synthase (iNOS); this in turn causes the production of arachidonic acid (AA) or nitric oxide (NO), respectively. Finally, the release of ATP via pannexin-1 (Panx1) hemichannels as well as of ATP and glutamate via P2X7Rs was also found to take place. The present data suggest that astroglial P2X7R stimulation is associated with neurological disorders leading to neuroinflammation, and apoptosis. The inset summarises examples of P2X7R mediated effects in astrocytes (artwork by courtesy of Dr. Jens Grosche)
P2X7Rs differentially modulate hippocampal and cortical astrocytes for example with respect to pore opening, p38 MAPK activation, caspase-1 activation and pro-IL-1β release [158]. These results open the possibility that activation of P2X7Rs influences the neuroinflammatory processes in distinct brain regions in different ways.
Recent studies identified several splice variants of the P2X7R that may account for the differential outcome of P2X7R activation in various experimental settings [159]. The splice variant P2X7A relates to pore formation and cell death, whereas the P2X7B variant does not induce plasma membrane permeabilisation and stimulates cell growth. All these data give evidence in favour of the versatility and plasticity of the P2X7R, and its ability to adopt different dispositions and conformations to elaborate particular actions when expressed natively in different cell types and tissues [160].
Intercellular signalling cascades: the P2Y (P2Y1) receptor
Four main heterotrimeric G protein subfamilies have been hitherto characterised (Gs, Gi/o, Gq/11 and G12/13) and individual P2YR subtypes may be linked to one or more of them [27, 30, 31, 102, 157]. The P2Y1, P2Y2, P2Y4, P2Y6 and P2Y11Rs couple to Gq proteins stimulating membrane-bound phospholipase C (PLC), which then cleaves phosphatidylinositol-bisphosphate (PIP2) in the membrane into two second messengers InsP3 and diacylglycerol (DAG). InsP3 mobilises [Ca2+]i, while DAG activates protein kinase C (PKC), leading to various other intracellular events, [27, 78, 157, 161]. P2Y12, P2Y13 and P2Y14Rs bind preferentially to the Gi/o subunit. Gi activation is classically associated with the inhibition of adenylate cyclase (AC) and reduce intracellular levels of cyclic adenosine-3′,5′-monophosphate (cAMP). The activation of the Gi/o subunit may also have other consequences [27, 31, 157].
P2YRs in astrocytes are linked to a variety of signal transduction mechanisms (Table 2 and Fig. 3). This receptor family can activate (1) the MAP kinase pathway, an obligatory step for the triggering and/or persistence of reactive astrogliosis, (2) the phosphoinositide 3-kinase (PI3K)/Akt-pathway, associated with cell growth, fibre regeneration, and inhibition of apoptosis and (3) the PKC pathway.

Schematic illustration of examples of signal transduction pathways in astroglial cells following P2Y1R activation. Stimulation of P2Y1Rs leads to the activation of phospholipases A2 and C (PLA2, C) and protein kinase C (PKC), as well as an increase in intracellular calcium ([Ca2+]i). The activation of P2Y1Rs result in the induction of second messenger and enzyme cascades, e.g. activation of the mitogen activated protein kinase (MAPK) pathway proteins (ERK1/2), p38 MAPK, c-Jun N-terminal kinase (JNK), and PI3K/Akt activation. P2Y1R-mediated signal transducer and activator of transcription 3 (STAT3) signalling may play a role in astrocyte proliferation and reactive astrogliosis. P2Y1R activation appeared to be involved in the activation of caspase (Casp) cascades and the release of arachidonic acid and increase in prostaglandin E2 (PGE2) levels. In addition, P2Y1R activation induces the activity of transcription factors such as nuclear factor κB (NF-κB), cyclic element-binding protein (CREB) and activator protein (AP-1) (which up regulate the expression of proinflammatory genes, e.g. c-Fos, c-Jun, c-Myc). Interaction between adenosine A1 and P2Y1Rs may alter the nucleotide signalling cascades. Modulation of astrocytic P2Y1Rs by the C-terminal domain of the gap junction protein connexin-43 (Cx43) appears to be involved in release of ATP and glutamate. The present data suggest that astroglial P2Y1R stimulation is associated with neurological disorders leading to neuroinflammation, and apoptosis. The inset summarises examples of P2Y1R-mediated effects in astrocytes (artwork by courtesy of Dr. Jens Grosche)
MAP kinases are a large protein family consisting of ERKs, the stress-activated protein kinases (SAPKs), the JNKs and p38/MAPKs, which are activated consecutively. The role of MAPK/ERKs is implicated in cell growth and differentiation, whereas SAPKs and p38 appear to regulate the cell death machinery. The importance of the ERK-cascade in mitogenic signalling by P2YRs in cultured astrocytes has been demonstrated repeatedly [162]. MAPKs are activated by phosphorylation at threonine and tyrosine residues [163] and phosphorylate themselves transcription factors, which control early gene expression, such as that of c-Fos [139, 164].
The involvement of MAPK/ERK in P2YR-mediated astrogliosis [165, 166], as well as caspase-3 activation and subsequent apoptosis [22, 167], have been described in vitro. The proliferation of astrocytes induced by traumatic injury in vivo and their modulation by pharmacological ligands suggest the involvement of the MAPK/ERK1/2- and PI3K/Akt-pathways in astroglial proliferation also in vivo [129].
ATP released by stretch-induced injury in vitro triggers ERK activation in astrocytic response to brain trauma [166]. Studies with P2R antagonists indicated a role for P2X2 and P2Y1Rs in injury-induced ERK-activation. P2Y2 [168] and P2X7Rs [169] are coupled to astroglial ERK. Sustained ERK1/2-activation is often associated with neuronal death, whereas acute ERK1/2-activation is typically a defence mechanism of cells in response to injury [170]. Stab wound and P2R stimulation by adenosine-5′-O-(2-thiodiphosphate) (ADPβS) caused ERK1/2-phosphorylation, which was inhibited by pretreatment with the P2X/YR antagonist pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS; [129]). After retinal detachment, the P2YR-mediated ERK1/2- and PI3 kinase-activation are necessary for mitogenic effects during proliferative vitreoretinopathy [171]. Weisman et al. [172] reported that proliferation of primary astrocytes by P2Y2R-stimulation depends on PI3K and ERK1/2.
The PI3K/Akt pathway is associated with controlling the balance between survival and cell death. Akt activation after trauma, axotomy and ischaemia is well established. The purinergic stimulation (e.g. P2X1,2,3, P2Y2Rs) of Akt phosphorylation in astrogliosis was studied in vitro (e.g. [22, 139, 173, 174]) and in vivo in the rat brain [129]. The involvement of P2Y1Rs in Akt activation in vivo suggests that the PI3K/Akt cascade stimulates astroglial proliferation and prevents apoptosis, indicating contribution of specific P2R subtypes during neurodegenerative diseases associated with excessive gliosis [129].
P2YRs may be coupled to phosphatidylinositol-specific phospholipase C (PI-PLC) via Gq and to ERK-signalling via Go or Gi. In cultured cortical astrocytes these receptors are coupled to the PI-PLC/calcium pathway [162]. Activation of the MAP kinase pathways can, in turn, activate transcription factors such as c-Fos, c-Jun, Elk1, ATF2 or the signal transducer and activator of transcription 3 (STAT3). Depending on the P2YR subtype, differences in the induction of signalling cascades were described. These signalling pathways may act synergistically for example by phosphorylating the immediate early gene products c-Fos and c-Jun, which both increase astrocyte proliferation and GFAP expression. c-Fos is an early gene product that contains in its promoter a binding site for the activating protein 1 (AP-1) and heterodimerizes to form an AP-1 complex [175–177]. Triple-labelling studies demonstrated c-Fos expression in P2Y1R-positive GFAP-labelled astrocytes (Fig. 4). Previous in vitro experiments reported an extracellular ATP-induced up-regulation of c-Fos protein and the formation of AP-1 transcriptional complexes [164, 178]. Additionally, PPADS reduced the incision-evoked c-Fos expression in a model of postoperative pain [179]. A role of the ERK-signalling cascade in gene regulation has intensively been studied [180, 181]. The activation kinetics of ERK signalling were associated with specific biological outcomes, whereas c-Fos was shown to function as a sensor for ERK1/2-signal duration; the use of the MAPK/ERK-inhibitor PD98059 suggest an ERK-mediated c-Fos expression [182].

Changes of c-Fos expression after stab wound injury in the NAc of rats. a Time course of c-Fos expression 2 h, 4 h, 24 h, 48 h and 4 days after ACSF- and ADPβS-microinjection in comparison to untreated controls (c) and plotted against log time (hours). b Schematic illustration of the localisation of the stab wound in the NAc of rats and the area (square) used for quantification of labelled cells (according to [127]; NAcc, core; NAcs, shell). c Quantification of the effects of ACSF, ADPβS (100 μM), PPADS (30 μM), PD98059 (50 μM; a MAPK inhibitor), wortmannin (Wo, 40 μM; a PI3K inhibitor), and the combination of ADPβS with the respective inhibitors, on the number of c-Fos-positive cells in the NAc of rats after a post-injection time of 2 h. The values are expressed as a percentage of ACSF-treated controls and represent the mean±SEM of five animals per group (*P<0.05, vs. ACSF group; + P<0.05, agonist ADPβS vs. agonist ADPβS/inhibitor group; # P<0.05, inhibitor vs. agonist/inhibitor group). d–g Confocal images of triple-immunofluorescence of d the P2Y1R subtype, e c-Fos-labelling and f GFAP-positivity (g, merge); thick arrow co-localisation with c-Fos; thin arrow no co-localisation with c-Fos. Scale bar: c–f = 20 μm
P2Y1,2,4 and P2X7Rs also couple to glycogen synthase kinase 3 (GSK3), a signalling molecule involved in cell survival, cell cycle regulation, proliferation and differentiation. The coupling of nucleotide receptors to GSK3 signalling was initially reported in cortical astrocytes [183]. P2Rs are coupled to GSK3β by a PKC-dependent pathway that is independent of Akt, p70 S6 kinase and the ERK pathways. These findings suggest that nucleotide signalling contributes to the regulation of GSK3 functions, one of which may be the response of astrocytes to CNS injury on the release of ATP [183].
P2X/YRs are also involved in the activation of NO signalling pathways and in the ATP/NO cross-talk. The presence of the Ca2+-dependent NO synthase (NOS) isoforms in astrocytes makes it conceivable that similarly to neurones, P2XR-mediated rises in Ca2+ may directly trigger NO production. Indeed, in purified cortical astrocytes in culture, ATP triggered the formation of NO through P2X2R-mediated [Ca2+]i elevation [184, 185].
Astroglial P2Y2Rs are coupled to αvβ3/β5 integrin signalling pathways, which control cytoskeletal remodelling and motility [172]. The activation of P2Y2 and P2Y4Rs regulates astrogliosis through ERK1/2 and STAT3 signalling cascade, mostly active in reactive astrocytes [35, 186]. P2Y2R activation induces the phosphorylation of the epidermal growth factor receptor (EGFR), response dependent upon the presence of a SH3 binding domain in the intracellular C terminus of the P2Y2R that promotes Src binding and transactivation of EGFR, e.g. regulating the proliferation of cortical astrocytes [187].
ATP triggers reactive astrogliosis by activation of a P2YR linked to the induction of cycloxygenase-2 (COX-2) [119, 188], and P2YR antagonists might counteract excessive COX-2 activation in both acute and chronic neurological diseases [188]. Transductional signalling to COX-2 induction involves an early activation of phospholipase A2 (PLA2) and arachidonic acid (AA) release; this may, hence, act as an inducer of the COX-2 gene via the PKC/MAPK pathway [113, 164, 189]. The different intracellular signalling pathways associated, e.g. with STAT3, nuclear factor κB (NF-κB), signalling molecules suppressor of cytokine signalling 3 (SOCS3), cAMP, JNK/c-Jun, FGF and MAPK are implicated in mediating different aspects and/or different degrees of reactive astrogliosis such as GFAP up-regulation, cell hypertrophy, proliferation and pro- or anti-inflammatory effects [for review see 130].
Astroglial response to CNS injury
Reactive astrogliosis and glial scar formation are prominent features of brain pathophysiology. However, the involvement of P2X/YRs was not characterised for all diseases, where astroglial activation was found (for example in metabolic disorders, infection, hepatic encephalopathy or Huntington’s disease). Here we present examples for several neurological diseases in relation to changes of astrocytes and nucleotide signalling. Some initial data on astroglial reaction in human tissue are also discussed (Table 3, Fig. 5).

Schematic diagram illustrating examples of P2X/YR expression in human brain on glial cells (especially astrocytes) in CNS neuropathological events (artwork by courtesy of Dr. Jens Grosche). For details, see also the Table 3
Acute CNS injury
Ischaemia/hypoxia
After hypoxia/ischaemia of the CNS, intracellular levels of ATP rapidly fall, whereas extracellular purine nucleotide concentrations are elevated due to cell damage [19–21, 190]. During in vitro ischaemia in hippocampal slices, ATP release occurred only after the anoxic depolarisation, whereas adenosine release was apparent almost immediately after the onset of ischaemia [191]. This suggests that ATP and adenosine release during ischaemia are independent processes with distinct underlying mechanisms, which consequently confer temporally distinct influences on neurones and glia in the ischaemic brain. Adenosine has an important protective role against ischaemic damage in the brain, possibly via A1 and A2R subtypes [89]. After all, these receptors are also able to influence P2YR activity, e.g. P2Y1R, as described above.
Ischaemic events result in membrane depolarisation and the release of glutamate and ATP that induces cytosolic calcium overload, mitochondrial damage and the activation of intracellular enzymes with deleterious consequences for cell function and viability. Ultimately, cell death and membrane damage cause additional massive release of glutamate and ATP, aggravating the initial damage [4, 9]. Ischaemic insults can open pannexin hemichannels and contribute to ATP release and the post-anoxic depolarisation in cultured neurones, although these results have not been confirmed in brain slices or in the in vivo animal studies [192]. ATP is hydrolysed by ATP diphosphohydrolase and 5′-nucleotidase; up-regulation of these enzymes after ischaemia has been reported and was considered as an indicator of an accelerated ATP release [19].
As frequently described, ischaemia may cause cellular degeneration and death, mediated through P2X and P2YRs [6, 88]. A post-ischaemic time- and region-dependent up-regulation of P2X (P2X2,4,7) and P2Y (P2Y1,2) receptors in neurones and glial cells was shown, suggesting the involvement of purinoceptors in the pathophysiology of cerebral ischaemia in vitro and in vivo.
In the cerebral cortex of spontaneously hypertensive rats, middle cerebral artery occlusion (MCAO) led to the up-regulation of P2X7R-immunoreactivity (IR) in the penumbra surrounding the necrotic region [193]. This IR was co-localised with astroglial and neuronal markers; immunoelectron microscopy revealed the expression of P2X7Rs on presynaptic elements of neurones and on glial cells. In order to clarify the mechanism of the ischaemic up-regulation, HEK293 cells were transfected with the recombinant human P2X7R, the C-terminus of which was labelled with EGFP; the transfected cells were afterwards subjected to in vitro hypoxia/ischaemia [194]. The combined use of confocal laser scanning microscopy, flow cytometry and electrophysiology suggested that 12 h of in vitro ischaemia increased the integration of intracellularly localised P2X7Rs into the plasma membrane of HEK293 cells. Neuronal changes observed under ischaemia [195, 196] may be due to an indirect effect via the P2X7R-induced release of an astrocytic, neuroactive signalling molecule (e.g. [197]), although direct effects at neurones cannot be excluded either [198, 199].
P2Y1Rs are strongly expressed in Purkinje cells, in the deep layers of the cerebral cortex and in ischaemia-sensitive areas of the hippocampus [200]. Astroglial P2Y1R expression and activity under ischaemic/hypoxic conditions possess a dual effect on cell survival/death, which depends on the extent of damage, the time course and levels of ATP release and the modulator influences of other cells. Intensive accumulation of P2Y1Rs was observed in the penumbra around the necrotic tissue [132]. In our own experiments, an increase in P2Y1R-IR was also found after mechanical injury in the peritraumatic area of the rat cortex and nucleus accumbens (NAc [100]) and after MCAO in the periinfarct region (H. Franke, U. Krügel and A. Lämmer, unpublished data). Additionally, using the MCAO model, we have demonstrated that the inhibition of P2Rs by PPADS improved the morphological and functional alterations induced by hypoxia/ischaemia [201]. In particular, the recovery of the cortical electrophysiological and motor functions after permanent MCAO in rats was considerably accelerated by PPADS. PPADS pretreatment also led to a decrease in the infarct volume and reduced cell death (neurones and astrocytes) for up to 7 days after MCAO compared to ACSF-treated controls [202]. In the periinfarct area, we have found the expression of P2X7Rs and observed a PPADS-sensitive up-regulation of P2Y1Rs (H. Franke, U. Krügel and A. Lämmer, unpublished data). In a rat model of cerebral ischaemia, the intracerebroventricular administration of the P2Y1R agonist MRS 2365 increased cerebral infarct volume 72 h after transient MCAO, whereas the P2Y1 antagonist MRS 2179 significantly reduced infarct volume and led to recovered motor coordination [132]. Furthermore, the P2Y1R agonist 2-methylthioladenosine 5′-diphosphate (2-MeSADP) has been reported to induce apoptosis by activating the human recombinant P2Y1R heterologously expressed in the 1321N1 astrocytoma cell line [167]. Otherwise, a significant cerebroprotection was observed with the P2Y1R selective antagonists MRS2279 and MRS2179, which allowed for the maintenance of motor coordination following transient MCAO [132].
P2Y1Rs in cultured astrocytes are involved in the protection against hydrogen peroxide (reactive oxygen species, generated in ischaemic stroke)-induced damage [136]. Data of Sun and co-workers [137] suggested that P2Y1Rs play a role in the production of GFAP and GDNF in astrocytes under transient MCAO and influence the related signalling pathways (Janus kinase 2/signal transducer [JAK2/STAT3], PI3K/Akt/CREB). Furthermore, it has been shown that after MCAO, inhibition of astrocytic P2Y1Rs results in cytokine/chemokine transcriptional suppression and cerebroprotection via a p-RelA-mediated NF-κB pathway [132].
Several studies have suggested roles for P2Y1Rs in the regulation of astrocyte swelling in particular due to an autocrine release of ATP from cultured astrocytes, (e.g. [136, 203]). Glial mitochondrial metabolism appears to be a key determinant of cytotoxic oedema and necrosis during cerebral ischemic stroke. The inhibition of glial mitochondria by ischaemia increases astrocyte swelling that leads to oncotic cell death [204]. P2Y1R stimulation reduces astrocyte cytotoxic oedema, necrosis and neuronal cell death in mice with Rose Bengal (RB)-induced brain lesions. In a RB-induced photothrombotic model of focal cerebral ischaemia, treatment with the P2Y1R agonist 2-MeSADP showed that the magnitude of ischaemic lesion was significantly reduced and the neurological outcome of oedema and cerebral ischaemic stroke was improved [204]. These data suggest that astrocyte mitochondria are a key energy source in the post-ischaemic penumbra. In fact, the mitochondrial metabolism can be stimulated by InsP3-mediated intracellular Ca2+ release, which significantly improves the neurological outcome of brain injuries [204]. This is in a good agreement with the finding that P2R-stimulated InsP3-mediated Ca2+ release enhances neuroprotection by an increase of the astrocytic mitochondrial metabolism during aging [205].
It should be noted that P2Y1Rs, found on the glia limitans and on vascular endothelium, are also important for ischaemic regulatory processes; for example, endothelial P2Y2R-mediated dilatations of rat middle cerebral artery in response to uridine-5′-triphosphate (UTP) were potentiated after ischaemia–reperfusion, while P2Y1R-mediated dilatation was simultaneously attenuated [206, 207]. Not only purine but also pyrimidine nucleotides appear to be released during ischaemia; the UTP-induced activation of P2Y4Rs causes cell death in human neuroblastoma cells [208].
An important question relates to the protective role of activated astrocytes and the P2Y1R expression in the penumbra after brain ischaemia. Li and co workers [41] showed in a GFAP−/−Vim−/− mice model that reactive astrocytes protect the penumbra during brain ischaemia. Mice deficient in both intermediate filament proteins, exhibited attenuated posttraumatic reactive astrogliosis. Seven days after MCA transection, infarct volume was considerably higher in GFAP−/−Vim−/− than in wild-type (WT) mice [41]. The absence of immunofluorescence for astrocytic markers was linked to changes in glutamate transporter, endothelin B receptor (ETBR)-mediated control of gap junctions, and plasminogen activator 1 (PAI-1) expression.
Mechanical trauma
Pathophysiological changes in traumatic brain injury (TBI) include cerebral ischaemia, oedema, release of excitatory neurotransmitters, oxidative stress, inflammation and apoptosis [36, 209]. Mechanical strain of rat cortical cultured astrocytes causes ATP release [22]; similarly, mechanical lesion of the brain increases extracellular ATP and glutamate [23].
A useful in vitro model for brain trauma was developed by Neary and co-workers (for review, see [139]). Astrocytes were cultured on deformable SILASTIC membranes and were subjected to rapid, reversible strain (stretch)-induced injury. These experiments demonstrated that ATP released by mechanical injury triggers ERK activation and suggested a role for extracellular ATP, P2Rs and calcium-dependent ERK signalling in astroglial responses to brain trauma [22, 166]. Studies with P2R antagonists indicated a role for P2X2 and P2Y1, but not P2X1, P2X3 or P2X7Rs for injury-induced astroglial ERK activation. The injury-induced activation of PKB/Akt in cultured astrocytes was also demonstrated [22]. Mechanical strain activates P2Y4Rs, which in turn stimulate the synthesis and release of thrombospondin-1, an extracellular matrix molecule that induces synaptogenesis during development and repair [141]. In another study, it was shown that P2Y2Rs promote the activation of ERK1/2 and PKB/Akt through PI3K dependent mechanisms after traumatic injury in astrocytic cells and enhance cell survival [174].
Astrogliosis in vivo was observed after direct infusion of ATP or ADP into one of the cerebral hemispheres, when compared with the contralateral hemisphere [210]. Intrastriatal microinjection of ATP causes P2XR-mediated cell death within 24 h in the striatum of rats, which supports the hypothesis that extracellular ATP mediates neuropathological events caused by brain injuries [211]. Finally, a single dose of the P2X/YR antagonist suramin injected intracerebrally through the needle used for producing a stab wound, reduced CNS inflammation, as judged by decreased cellular proliferation, GFAP levels and tenascin C-IR [212]. In addition, examination of the corpus callosum of saline-treated animals 30 days post-trauma revealed a disruption of the fibre tract within the lesion site, while suramin-treated animals displayed numerous fibres spanning the lesion. These results demonstrate that a single injection of suramin transiently inhibits the astrogliotic response, which may be sufficient to ameliorate subsequent tissue damage [212].
We have demonstrated that endogenous ATP participates in the in vivo astrogliosis caused by stab wound injury in the NAc and cortex of rats [127]; and we have ound an increase in GFAP and astroglial proliferation following microinjection of ATP analogues into the NAc [127, 128]. P2X and P2Y receptors were expressed/co-expressed in astrocytes of untreated rats; the P2R expression (P2X1,7 and P2Y2,6) was up-regulated by mechanical injury [76, 100, 213]. Pharmacological characterisation showed that astroglial activation was mediated specifically via P2Y1Rs (Fig. 1G–I) [100, 128, 129]. Additionally, in this stab wound, paradigm P2Y1R activation enhanced the early apoptosis marker caspase-3 in a PPADS reversible manner. Pretreatment with PPADS prevented the P2Y1R mediated rise of the apoptotic pERK1/2 and the anti-apoptotic pAkt [129]. Our data indicated that traumatic injury in vivo is accompanied by a P2Y1R-mediated modulation of PI3K/Akt- and MAPK/ERK-signalling pathways in astrocytes and neurones, resulting in astroglial proliferation and anti-apoptotic processes [129].
Finally, we also demonstrated the expression of the P2Y1Rs in activated astrocytes in the pericontusion area after TBI in the prefrontal cortex of the human brain (post-mortem autopsy material, Fig. 1J–L [214]).
Neurodegenerative diseases
Alzheimer’s disease
Astrocytes become activated in Alzheimer’s disease (AD) and their accumulation has been observed around amyloid deposits in the brain, suggesting a role of these cells in the homeostasis of amyloid plaques and in modulating the neuronal environment in AD [34, 45].
The elevated levels of pro-inflammatory cytokines (e.g. IL-1β) can enhance the non-amyloidogenic amyloid precursor protein (APP) processing through up-regulation of the P2Y2Rs in neurones, as shown in primary cortical neuronal cell cultures [215]. The activation of P2Y2Rs by UTP appears to be neuroprotective, since this nucleotide enhanced the non-amyloidogenic cleavage of APP [216]. Recent data obtained in primary cultures of rat cortical astrocytes indicate that APP production and secretion can be regulated by activation of heteromeric P2Y2/4Rs coupled to protein kinase signalling pathways (MAPKs, but not Akt) and suggest that astrocytes can be a potential source of APP [217]. In conclusion, astrocytic P2Y2R activation regulates a variety of signal transduction pathways, such as (1) PLC, activating different second messenger cascades (e.g. PKC, PLA2, ERK1/2); (2) phosphorylation of the EGFR, depending on the direct interaction of a Src homology binding sequence, promoting the transactivation of EGFR and the regulation of some key functions (e.g. the proliferation of cortical astrocytes); and (3) activation of αVβ3/5 integrins that bind directly to an Arg-Gly-Asp (RGD) motif in the first extracellular loop of the P2Y2R subtype, mediating cytoskeletal rearrangements and increasing astrocyte chemotactic activity, finally associated with reactive astrogliosis in AD brains [187, 218]. It should be noted that ATP and UTP in their interaction with the “sensor” P2Y2R have been identified as critical “find-me” signals important for phagocyte chemoattraction by apoptotic cells [219]. Peterson and co workers [187] summarise the mechanisms underlying the pro-inflammatory and neuroprotective effects mediated by P2Y2Rs in glial cells and neurones and their potential relationship to the pathophysiology of AD. The main hypothesis is that chronic stress or CNS damage causes an increased nucleotide release from astrocytes and the induction of pro-inflammatory responses in glial cells with subsequent up-regulation of neuronal P2Y2Rs, which in turn mediate neuroprotective effects. The involvement of P2Y2Rs in microglia during inflammatory processes was also emphasised. Recent data using brain slices indicated that activation of P2Y2Rs with UTP results in an inflammatory response, measured as a rapid rise in intracellular Ca2+ and release of cytokines [220].
In mouse primary astrocytes, the specific activation of P2X7Rs induced the release of the fragment soluble (s)APPα [221]. The capacity of P2X7Rs to trigger non-amyloidogenic APP processing may tentatively suggest that astrocytes have a neuroprotective effect in AD. It was demonstrated that the MAP-kinase modules ERK1/2 and JNK are involved in P2X7R-dependent α-secretase activity. Extracellular ATP, acting through P2X7Rs, can alter β-amyloid peptide (Aβ)-induced cytokine release from macrophages and microglia and makes this receptor-subtype to an important modulator of neuroinflammation in AD [222, 223]. Semiquantitative RT-PCR showed enhanced expression of P2X7R mRNA in AD microglia in comparison with microglia from non-demented brains [224]. Immunohistochemical analysis showed prominent P2X7R expression in association with Aβ plaques and a localisation to HLA-DR (class II major histocompatibility antigen)-immunoreactive microglia.
A possible role of P2YR alteration in regulating molecular processes, which underlie plaque formation was investigated using post-mortem materials from a cohort of longitudinally assessed AD patients and aged controls [225]. Interestingly, the receptor-deficit seems to be subtype-specific and was manifested by reduction in P2Y2Rs, while P2Y4 and P2Y6Rs remained unchanged. P2Y2R-IR was observed to be selectively reduced in the AD parietal cortex correlating with neuritic plaque and neurofibrillary tangle scores as well as synapse loss. Moreover, decreases in parietal P2Y2R densities also correlated with lower synaptophysin-IR, a marker of synapse loss [225]. In addition to changes of the P2Y2R expression in AD, alterations in the density of the P2Y1R subtype [226] as well as prominent changes in the number of the P2X7Rs (e.g. [227] were also observed. P2X7Rs are up-regulated around senile plaques in animal models of AD and in patients with AD [224, 227]. P2Y1Rs have been immunhistochemically localised at neuritic plaques and neurofibrillary tangles [226].
Parkinson’s disease
In autopsies of the substantia nigra and putamen from Parkinson’s disease (PD) patients, reactive astrogliosis is generally mild or moderate [228]. Astrocytes have been implicated as potentially exerting both neurotoxic and neuroprotective activities in PD [36].
Endogenous adenosine and ATP appear to modulate dopaminergic neurotransmission in the CNS. Exogenously applied ATP enhances the release of dopamine from the terminals of neurones projecting from the substantia nigra pars compacta to the striatum via P2YRs [229] and modulate the release of dopamine from the respective axon terminals in the NAc [230–232]. The tonic dopamine release may be physiologically regulated by P2YRs [231, 232]. The P2R-IR in the nigro-striatal and mesocortico-limbic dopaminergic systems ex vivo and in vivo has been documented [231, 233], as well as a trophic growth promoting effect of P2R agonists on fibre outgrowth in the dopaminergic projection system [233].
In an animal model of PD using 6-hydroxydopamine-lesioned substantia nigra, the expression of the P2X7R in nigral microglial and astroglial cells was observed, but was absent in dopaminergic neurones. The enhanced neuroinflammatory process through the activation of microglia and the release of inflammatory agents was discussed in this context [234].
In the PD, the main focus of research is concentrating on striatal A2ARs being a target of adenosine generated by the enzymatic degradation of synaptically released ATP [235, 236]. Specific A2AR antagonists are currently being investigated for the treatment of the PD. However, neuronal rather than astrocytic A2ARs appear to be drug targets for this purpose and are therefore not discussed here in detail.
Multiple sclerosis and amyotrophic lateral sclerosis
In human multiple sclerosis (MS) lesions of autopsy brain tissues, immunoreactivity for the P2X7R was demonstrated on reactive astrocytes [237]. In cultures of primary human foetal astrocytes, P2X7R stimulation increased the production of IL-1-induced NOS. These studies suggest that signalling via the P2X7R may modulate the astrocytic response to inflammation [237]. An immunofluorescence and confocal analysis in sections of cerebral cortex from postmortem MS brain indicated that P2Y12 protein is present in myelin and in interlaminar astrocytes; P2Y12Rs were also highly enriched in oligodendrocytes [238].
Furthermore, astrocytes (via P2X7Rs), infiltrating cells of the monocyte/macrophage lineage and activated microglia can release ATP (possible through P2X7R pores; [239]) and thus contribute to further P2X7Rs activation and cell death in oligodendrocytes [192, 240]. An increased expression of P2X7Rs in axon tracts before lesions are formed indicating that this feature may constitute a risk factor associated with newly forming lesions in MS; the P2X7R may thus prove to be a diagnostic and/or prognostic clinical biomarker for MS [192].
Finally, the P2Y-like receptor GPR17 has been identified as a modulator of oligodendrocyte differentiation during maturation and in myelinating disorders. In primary cultures of astrocytes and oligodendrocyte precursor cells (OPCs), the GPR17 expression was interpreted as a “danger signal” in the presence of high extracellular ATP concentrations, suggesting an important role of the receptor in brain disorders [241, 242].
Amyotrophic lateral sclerosis (ALS) is a neuromuscular disease causing progressive paralysis of all voluntary muscles. A pathological hallmark of ALS is neuroinflammation mediated by non-neuronal cells in the nervous system, such as microglia and astrocytes that accelerate the disease progression [243]. Recent data suggest that during the progression of ALS, microglia, astrocytes and motor neurones might cross-talk via ATP release/degradation and P2X7R activation, generating a feedback loop that drives a sustained pro-inflammatory and detrimental response [243, 244]. In ALS, extracellular ATP and P2X7Rs are involved in astrocyte-mediated motor neurone death [245]. Mutant superoxide dismutase 1-positive astrocytes displayed increased ATP-dependent proliferation and basal increase in extracellular ATP degradation. The pharmacological inhibition of P2X7Rs (e.g. by Brilliant Blue G) might reduce neuroinflammation in ALS through astrocytes [245].
Epilepsy
Epilepsy, characterised by recurrent unprovoked abnormal excessive or synchronous neuronal activity, is often accompanied by reactive astrogliosis [246, 247]. Glial alterations may contribute to the generation and spread of seizure activity. Recently astroglial Ca2+ waves have been shown to provoke seizures, and conventional antiepileptic drugs were able to attenuate these Ca2+ waves [65, 89, 192, 236, 248, 249].
Microinjection of ATP analogues into the prepiriform cortex induces generalised motor seizures [236]. Blocking ATP-mediated gliotransmission, therefore, represents a potential target for antiepileptic drugs. Adenosine is an endogenous inhibitor of excitatory synaptic transmission with potent anticonvulsive properties. Astrocytes are considered as the main regulators of adenosine release, which acts as the brain’s natural anticonvulsant, and the basal synaptic levels are controlled by an astrocyte-based adenosine cycle in which a key element is the activity of adenosine kinase (ADK; “ADK hypothesis of epileptogenesis”) [192, 250]. Adenosine accumulates from the hydrolysis of ATP (by ectonucleotidases) released by astrocytes and is believed to inhibit distant synapses by acting on adenosine P1 receptors [89, 192, 248].
Other potential roles for ATP signalling in the initiation and spread of epileptiform discharges may involve synaptic plasticity and coordination of synaptic networks [248]. Adenosine functions are consistent with a surround-inhibitory mechanism whose failure would predispose to seizures. Glutamate and extracellular ATP can activate astrocytes and trigger calcium signals modulating local excitability [68]. Glutamate exocytosis from astrocytes of the rat hippocampal dentate molecular layer enhances synaptic strength at excitatory synapses between perforant path afferents and granule cells. The NMDA receptor 2B subunits on the extrasynaptic portion of excitatory nerve terminals are spatially related to glutamate-containing synaptic-like microvesicles in the apposed astrocytic processes. This glial regulatory pathway is endogenously activated by neuronal activity-dependent stimulation of astrocytic P2Y1Rs as an evidence for a physiological control of synaptic activity via exocytosis of glutamate from astrocytes [68]. UTP, which is released during epileptic activity in the 4-aminopyridine-induced epilepsy model acts as an inhibitory neuromodulator by stimulating P2YRs [251].
Enhanced P2X7-IR accompanied with mossy fiber sprouting at the dentate gyrus of epileptic animals, suggesting a role of nucleotide receptors in the pathophysiology of temporal lobe epilepsy. Otherwise, a decrease in presynaptic P2XRs in the hippocampus of rats after kainate-induced convulsions might be associated with the development of seizures and/or neurodegeneration during epilepsy (reviewed in [89]).
Increased expression of P2X7Rs was found in the hippocampus of chronic epileptic rats which show enhanced responses to ATP [252]. After a status epilepticus (SE), an increased expression of P2X7, P2Y6, P2Y12Rs mRNAs in the hippocampus has been observed [253]. Significantly elevated P2X7R-IR in amoeboid or phagocytic microglia appeared in the dentate gyrus 7 days after SE and at this time point the loss of GFAP-IR was very pronounced, indicating that the network of astrocytes was disrupted [149]. The treatment with PPADS and suramin markedly, but not completely, inhibited microglial activation following SE and suggest that astroglial death induced by activated microglia may be pathophysiologically relevant for epileptogenesis [149].
Finally, the variation in ATP, ADP and AMP hydrolysis rates and of the soluble nucleotide phosphodiesterase (PDEase) activity in the serum of patients suffering from epilepsy was investigated, and an increase in nucleotide hydrolysis was reported [254]. It was hypothesised that the higher nucleotidase activity might play a role in the modulation of epileptic events.
Chronic exposure to drugs
Ethanol
Chronic treatment with ethanol induces astrogliosis in rats (e.g. [255, 256]). The influence of ethanol on P2R-mediated PLD activity in primary astroglial cultures was postulated, since ADP was almost as efficient as ATP in inducing phosphatidylethanol formation, whereas AMP was significantly less potent [257]. Using cultured astrocytes, it was found that treatment with ethanol affects glutamate uptake but not adenosine uptake by affecting PKC modulation of transporter activity [258]. Moreover, the apoptotic effect of ethanol was potentiated by caffeine-induced calcium release [259], suggesting a correlation between cytosolic Ca2+ increase and the rate of apoptosis.
Acute exposure to ethanol elevates extracellular adenosine levels by selectively inhibiting the type 1 equilibrative nucleoside transporter (ENT1; [260]). Pharmacological inhibition or genetic deletion of ENT1 reduces the expression of excitatory amino acid transporter 2 (EAAT2), the primary regulator of extracellular glutamate concentrations in astrocytes. These data support a central role for adenosine-mediated glutamate signalling and the involvement of astrocytes in regulating ethanol intoxication and preference.
Amphetamine
Dysregulation in the purinergic, dopaminergic and glutamatergic systems are possible causes for schizophrenia, PD or drug addiction. As described in “Parkinson’s disease”, extracellular ATP acting via P2YRs enhances dopamine release in the striatum and NAc. We have found that P2Y1Rs are up-regulated in astrocytes of the striatum and NAc of rats treated with amphetamine (AMPH). Re-application of AMPH resulted in behavioural sensitisation and astrogliosis [261]. PPADS partly prevented the AMPH-induced astrogliosis as well as the up-regulation of P2Y1 receptors and inhibited the development of sensitisation.
The expression of inducible NOS (iNOS) after AMPH-treatment was also observed and its inhibition after PPADS pretreatment suggests a link between P2Y1R-induced astrogliosis, neuronal reactions and behavioural changes. The NO formation could be prevented by suramin and PPADS. The hypothesised interaction between NO and P2Y1Rs was supported by the fact that the blockade of P2Rs by suramin or oxidised ATP (oxATP) in astrocytes resulted in inhibition of the NOS accumulation and the down-regulation of NO production [262]. In our own studies, we found a cellular co-localisation of P2Y1Rs and iNOS, suggesting that astrocytes, expressing nucleotide receptors, may act as a link between the ATP-mediated P2Y receptor activation, NOS stimulation and NO induced up-regulation of cGMP [261, 263].
Neuropathic pain
The plasticity of spinal cord neurones and of the primary afferent nerves relaying peripheral information to the spinal cord was suggested to be the cause of chronic pain [89]. The occurrence of spinal cord astrogliosis in most pathological pain models is well known. Garrison et al. [264] used the sciatic constrictive injury (CCI) neuropathic pain model and found a strong correlation between elevated GFAP levels in the spinal cord dorsal horn and neuropathic pain measured in the same animals. In various types of neuropathic and inflammatory pain models, spinal astrogliosis usually spreading to all spinal cord lamina is described [265]. Furthermore, the degree of astrogliosis appeared to follow the increase in microglia numbers, although astrogliosis as well as hyperalgesia outlast the up-regulation of microglia [266]. These findings suggest that astrocytes are involved in pain modulation and pain processing.
ATP is known to be of major importance for the initiation and maintenance of pain symptoms [267]. It appears that astrocytes play critical roles in pathological pain (1) through the secretion of diffusible transmitters (e.g. interleukins, ATP, TNF, PGE and NO); (2) by augmenting primary afferent neuronal signalling or sensitising second order neurones in the spinal cord; (3) by directly modulating synaptic transmission between neurones in the nociceptive pathway; and (4) by creating astrocyte networks that convey signals across and along the spinal cord [36, 265, 268].
ATP release from Ca2+ wave propagating spinal astrocytes could play an important role in the conduction of nociceptive information, since the central terminals of dorsal root ganglion (DRG) neurones have been found to respond to ATP by depolarisation and glutamate release [269]. The amount of ATP secreted by astrocytes in response to glutamate stimulation in vitro was increased by substance P in a concentration-dependent manner [270], thus indicating a role for astrocytes in facilitating nociceptive transmission from peptidergic nociceptors.
The peripheral equivalents of astrocytes, the so-called satellite glial cells located in sensory ganglia, bear functional P2X7Rs [271]. Thus, it was suggested that ATP released from DRG neurones may activate P2X7Rs at satellite cells, which in consequence release TNF-α that potentiates P2X3R-mediated responses at nearby neurones thereby regulating pain sensation [272]. Thus, DRG neurones and surrounding glial cells communicate with each other via signalling molecules; this communication has implications for the manifestation of chronic pain [273].
Furthermore, a role for astrocytes in pain modulation is supported by the therapeutic effects of fluorocitrate treatment, which specifically blocks astrocyte metabolism, thus revealing astrocytes as potential drug targets in clinical pain management [265]. Future research in astrocyte activation and signalling may therefore reveal novel drug targets for managing pathological pain.
Toxicology
Lead (Pb)
Exposure of the immature rat brain to lead (Pb), a potent neurotoxin, resulted in regional changes in the concentration of purines and selected nucleotide receptors [274]. In all examined brain structures (cortex, cerebellum, hippocampus), a decrease in ATP and ADP levels was found. Furthermore, an increase in A1R and adenosine concentrations suggests protective mechanisms mediated by adenosine. In the hippocampus, most vulnerable to Pb, an enhanced expression of P2X7R and connexin-43 in the astroglia was found, suggesting the involvement of astrocytes in pathophysiological changes observed after Pb-exposure in immature rat brain.
Trimethyltin
Trimethyltin (TMT) is a compound that generates neurotoxic effects and is used to model AD lesions in the brain. TMT causes astrogliosis and neuronal death in the hippocampus and other limbic regions [185]. These authors examined the expression of nitric oxide synthase (NOS) and P2XRs (P2X1,2,4,7) that were induced by TMT administration at different time points. Massive glial activation and neuronal death in the CA1 and CA3 regions were observed after TMT treatment. Furthermore, expression of astrocytic P2X2R and neuronal NOS were temporarily enhanced in association with the progression of neuronal death. TMT-induced hippocampal degeneration is associated with nitrergic and nucleotide activations. In this model, the two systems were active in segregated cell populations, namely, P2XRs in astrocytes and NOS in neurones. Finally, the temporal relations between P2XR and NOS expression and neuronal degeneration suggest interactions between P2XR/NO signalling and cell survival.
Glioma
Alterations in nucleotide signalling are involved in tumour generation [275, 276]. Glioblastomas are the most common form of primary tumours of the CNS and despite treatment, patients with these tumours have a very poor prognosis. Glioma C6 are chemically transformed astrocytes, often used as an experimental model system for studying glioblastoma growth and invasion [277–279]. However, glioma cell lines may not represent all the phenotypic and genetic characteristics of the primary tumours [280].
Recent data suggest that nucleotide signalling is involved in the growth and progression of gliomas [281]. ATP plays an important role in glioma proliferation in vivo and glioblastoma growth is reduced by the ectonucleotidase apyrase, indicating the role of nucleotide mechanisms in tumour development [282]. Glioma cell lines have altered extracellular ATP, ADP, and AMP catabolism, showing low rates of extracellular ATP hydrolysis and high rates of extracellular AMP hydrolysis when compared to primary astrocytes in culture [283]. ATP accumulation in the tumour periphery results in glioma cell proliferation and neuronal toxicity. Cell lysis caused by excitotoxic death or by tumour resection may liberate intracellular ATP, a known mitotic factor for glioma cells [277]. Adenine nucleotides induce cell proliferation in diverse human glioma cell lines and the majority of glioma cell lines are resistant to cell death induced by cytotoxic ATP concentrations [277, 284]. Morrone and co-worker [277] showed that the glioma cells exhibit resistance to death induced by ATP, when compared with a normal tissue. Indeed, high ATP concentrations induced astrocytic death after 24 h in organotypic cell cultures of the brain but not in glioma cell lines. In conclusion, ATP released in these situations can induce cell death of the normal tissue surrounding the tumour, potentially opening space for the fast growth and invasion of the tumour tissue [277].
The effects of nucleotides on the growth of C6 glioma cells are regulated by ectonucleotidases, e.g. ecto-nucleotide pyrophosphatase (ecto-NPP) and by the activation of nucleotide receptors [285, 286]. Gliomas have low expression of all ecto-nucleotide diphosphohydrolases (E-NTPDases), particularly NTPDase2, when compared to astrocytes in culture. It has been shown that selective NTPDase2 expression modulates in vivo rat glioma growth, specifically, ADP derived from NTPDase2 activity stimulates platelet migration to the tumour area and NTPDase2, by regulating angiogenesis and inflammation, seems to play an important role in tumour progression [281].
A number of studies were performed in human astrocytoma cell lines. Analysis of the C6 glioma cell line and C6 ex vivo glioma cultures revealed distinct cell morphologies, although cell differentiation and angiogenic marker expressions were similar [280]. With regard to nucleotide receptor expression, it has been found that both the C6 cells and the C6 ex vivo glioma cultures express purinoceptors including P2X4, P2Y1, P2Y2, P2Y4, P2Y12, P2Y13 and P2Y14 [280]. In addition, rat C6 glioma cell line and C6 ex vivo glioma cultures exhibited similar extracellular ATP metabolism and cell proliferation behaviour when exposed to cytotoxic ATP concentrations.
The P2Y1R has a dual role; it has been implicated in decreased cell proliferation in different forms of cancer (e.g. melanomas) but it is also a cell proliferation stimulus in glioma cells [287, 288]. P2Y1Rs coupled to PLC are responsible for the release of [Ca2+]i. P2Y2Rs are related to increased cell proliferation by activation of the PLC pathway, and P2Y12Rs are negatively coupled to AC [276, 278, 287, 289]. P2Y2R-mediated proliferation through the Ras/ERK-pathway as well as differential effects of the P2Y1 and P2Y12 receptor subtypes on ERK1/2, MAPK and PI3K signalling and cell proliferation in serum-deprived and non-starved glioma C6 cells were repeatedly reported [278, 287, 290]. In addition, the P2Y6R activates PKC to protect 1321N1 astrocytoma cells against TNF-induced apoptosis [291].
Of interest are data indicating that P2Y1, P2Y2 and P2Y12Rs can exist on intracellular membranes of mitochondria in rat astrocytes and glioma C6 cells [279]. Their role in these sub-cellular structures remains unclear. Expression and functional characterisation of nucleotide receptors in long-term serum-deprived glioma C6 cells showed an increased expression of P2Y12Rs relative to P2Y1Rs in relation to a reduced ERK1/2 phosphorylation and an enhanced Akt-phosphorylation [292]. Akt phosphorylation increased in parallel with the low expression of the P2Y1R, indicating an inhibitory role of P2Y1 in Akt pathway signalling. The reported shift in nucleotide receptor expression from P2Y1 to P2Y12 is supposed to be a new and important self-regulating mechanism that promotes cell growth rather than differentiation and is a defence mechanism against effects of serum deprivation [292]. Finally, thrombin-promoted release of UDP-glucose from human 1321N1 astrocytoma cells, and the resulting activation of P2Y14Rs are interesting observations [293]. Given the presence of P2Y14 receptors in astrocytes, UDP-glucose may have important autocrine/paracrine functions in the brain at least under pathological conditions.
Immunohistochemical analysis of P2X4R expression in the rat C6 glioma indicated specific expression in compact and infiltrative tumour growth areas, observed in a distinct subset of tumour-associated macrophages and activated microglia in glioma, whereas at GFAP- and nestin-immunopositive astrocytes P2X4Rs were hardly found [294]. Interestingly, P2X7Rs, which mediate cell apoptosis, are absent in both the C6 cell line and the C6 ex vivo glioma cultures [280]. Finally, cytotoxic ATP concentrations do not alter C6 ex vivo glioma cell proliferation [277], in agreement with the previously described absence of the P2X7Rs [280, 295].
Conclusions
Astrocytes have important roles in the CNS supporting neurones during normal and pathological conditions. In neural injury caused by all types of traumatic, metabolic, hereditary and infectious diseases, astrocytic reactions are survivalistic and beneficial, but in case of excessive pathological stress, they turn into deleterious, aggravating the outcome of the insult.
Reactive astrogliosis and glial scar formation are activated by brain lesions both acute and chronic and are prominent factors in neurodegenerative diseases such as Alzheimer’ disease, Parkinson’s disease, multiple sclerosis and amyotrophic lateral sclerosis, and epilepsy-induced secondary neurodegeneration. Astrogliosis can be also induced by consumption of addictive drugs or certain neurotoxins such as lead or trimethylin. The involvement of astrocytes in neuropathic pain is in the meantime also firmly established.
It is quite natural that in view of the plethora of neurological diseases in which extracellular nucleotides may be a pathological factor, P2X and P2Y receptor antagonists are considered to be possible therapeutic drugs. The finding of P2X7Rs as critical mediators of neuronal and glial death in neurodegeneration of grey and white matter points to these receptors as key targets for the development of neuroprotective drugs. Hence, a lot of energy was brought up to synthesise new P2X7R antagonists with high selectivity and improved bioavailability. These compounds belong to a series of chemical classes (e.g. benzamides, bicycloheteroaryls, acylhydrazines, biaromatics, and protoberberines). The considerable activities in academia and pharmaceutical companies with the aim of introducing new therapeutically applicable P2X7R antagonists underline the great need to use these compounds as an adjunct, non-causal therapy for the CNS illnesses discussed above. Hopefully, these efforts will lead to a major breakthrough in the near future.
Abbreviations
- AA:
-
Arachidonic acid
- Aβ:
-
Oligomeric β-amyloid peptide
- AC:
-
Adenylate cyclase
- AD:
-
Alzheimer’s disease
- AKT:
-
Serine-threonine kinase AKT
- ALS:
-
Amyotrophic lateral sclerosis
- APP:
-
Amyloid precursor protein
- ATP:
-
Adenosine 5′-triphosphate
- BrdU:
-
5-Bromo-2′-deoxyuridine
- [Ca2+]i :
-
Intracellular free calcium concentration
- cAMP:
-
Cyclic adenosine-3′,5′-monophosphate
- cGMP:
-
Cyclic guanosine-3′,5′-monophosphate
- CNS:
-
Central nervous system
- COX:
-
Cyclooxygenase
- DAG:
-
Diacylglycerol
- DRG:
-
Dorsal root ganglion
- EGF:
-
Epidermal growth factor
- EGFP:
-
Enhanced green fluorescent protein
- ERK:
-
Extracellular signal regulated protein kinase
- FGF:
-
Fibroblast growth factor
- GFAP:
-
Glial fibrillary acidic protein
- GSK3:
-
Glycogen synthase kinase 3
- IL:
-
Interleukin
- InsP3 :
-
Inositol (1,4,5)-trisphosphate
- IR:
-
Immunoreactivity
- JNK:
-
Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MCAO:
-
Middle cerebral artery occlusion
- MS:
-
Multiple sclerosis
- NAc:
-
Nucleus accumbens
- NGF:
-
Nerve growth factor
- NG2:
-
Chondroitin sulphate proteoglycan
- NF-κB:
-
Nuclear factor-κB
- NO:
-
Nitric oxide
- PD:
-
Parkinson’s disease
- PDGF:
-
Platelet-derived growth factor
- PGE2 :
-
Prostaglandin E2
- PKC:
-
Protein kinase C
- PI3K:
-
Phosphatidylinositol 3-kinase
- PL(A2):
-
Phospholipase (A2)
- PPADS:
-
Pyridoxal-phosphate-6-azophenyl-2′,4′-disulphonic acid
- P2R:
-
Purinergic receptor
- SAPK:
-
Stress-activated protein kinase
- SE:
-
Status epilepticus
- STAT3:
-
Signal transducer and activator of transcription 3
- TBI:
-
Traumatic brain injury
- TNF:
-
Tumor necrosis factor
- UTP:
-
Uridine 5′-triphosphate
References
Butt AM, Hamilton N, Hubbard P et al (2005) Synantocytes: the fifth element. J Anat 207:695–706
Kettenmann H, Ransom BR (2005) Neuroglia. Oxford University Press, Oxford, p. 601
Verkhratsky A, Butt A (2007) Glial neurobiology. A textbook. Wiley, Chichester, p 220
Franke H, Illes P (2006) Involvement of P2 receptors in the growth and survival of neurons in the CNS. Pharmacol Ther 109:297–324
Burnstock G (2007) Physiology and pathophysiology of purinergic neurotransmission. Physiol Rev 87:659–797
Burnstock G (2008) Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 7:575–590
Burnstock G, Verkhratsky A (2009) Evolutionary origins of the purinergic signalling system. Acta Physiol (Oxf) 195:415–447
Neary JT, Zimmermann H (2009) Trophic functions of nucleotides in the central nervous system. Trends Neurosci 32:189–198
Verkhratsky A, Krishtal OA, Burnstock G (2009) Purinoceptors on neuroglia. Mol Neurobiol 39:190–208
Illes P, Verkhratsky A, Burnstock G et al (2012) P2X Receptors and their roles in astroglia in the central and peripheral nervous system. Neuroscientist. doi:10.1177/1073858411418524
Tozaki-Saitoh H, Tsuda M, Inoue K (2011) Role of purinergic receptors in CNS function and neuroprotection. Adv Pharmacol 61:495–528
Zimmermann H (2011) Purinergic signaling in neural development. Semin Cell Dev Biol 22:194–204
North RA, Verkhratsky A (2006) Purinergic transmission in the central nervous system. Pflüger’s Arch 452:479–485
Pankratov Y, Lalo U, Verkhratsky A et al (2006) Vesicular release of ATP at central synapses. Pflüger’s Arch 452:589–597
Pankratov Y, Lalo U, Verkhratsky A et al (2007) Quantal release of ATP in mouse cortex. J Gen Physiol 129:257–265
Araque A, Navarrete M (2010) Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci 365:2375–2381
Lalo U, Verkhratsky A, Pankratov Y (2011) Ionotropic ATP receptors in neuronal-glial communication. Semin Cell Dev Biol 22:220–228
Hamilton NB, Attwell D (2010) Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11:227–238
Braun N, Zhu Y, Krieglstein J et al (1998) Upregulation of the enzyme chain hydrolyzing extracellular ATP after transient forebrain ischemia in the rat. J Neurosci 18:4891–4900
Juranyi Z, Sperlagh B, Vizi ES (1999) Involvement of P2 purinoceptors and the nitric oxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res 823:183–190
Melani A, Turchi D, Vannucchi MG et al (2005) ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int 47:442–448
Neary JT, Kang Y, Tran M et al (2005) Traumatic injury activates protein kinase B/Akt in cultured astrocytes: role of extracellular ATP and P2 purinergic receptors. J Neurotrauma 22:491–500
Franke H, Grummich B, Härtig W et al (2006) Changes in purinergic signaling after cerebral injury–involvement of glutamatergic mechanisms? Int J Dev Neurosci 24:123–132
Zimmermann H, Braun N (1999) Ecto-nucleotidases—molecular structures, catalytic properties, and functional roles in the nervous system. Prog Brain Res 120:371–385
Zimmermann H (2006) Ectonucleotidases in the nervous system. Novartis Found Symp 276:113–128
Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50:413–492
Abbracchio MP, Burnstock G, Boeynaems JM et al (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442:527–532
Abbracchio MP, Burnstock G, Verkhratsky A et al (2009) Purinergic signalling in the nervous system: an overview. Trends Neurosci 32:19–29
von Kügelgen I, Harden TK (2011) Molecular pharmacology, physiology, and structure of the P2Y receptors. Adv Pharmacol 61:373–415
Köles L, Leichsenring A, Rubini P et al (2011) P2 receptor signaling in neurons and glial cells of the central nervous system. Adv Pharmacol 61:441–493
Giaume C, Kirchhoff F, Matute C et al (2007) Glia: the fulcrum of brain diseases. Cell Death Differ 14:1324–1335
Rodriguez JJ, Olabarria M, Chvatal A et al (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16:378–385
Heneka MT, Rodriguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration. Brain Res Rev 63:189–211
Robel S, Berninger B, Götz M (2011) The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci 12:88–104
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Butt AM (2011) ATP: a ubiquitous gliotransmitter integrating neuron–glial networks. Semin Cell Dev Biol 22:205–213
Verkhratsky A (2010) Physiology of neuronal-glial networking. Neurochem Int 57:332–343
Verkhratsky A, Parpura V, Rodriguez JJ (2011) Where the thoughts dwell: the physiology of neuronal-glial “diffuse neural net”. Brain Res Rev 66:133–151
Freeman MR (2010) Specification and morphogenesis of astrocytes. Science 330:774–778
Li L, Lundkvist A, Andersson D et al (2008) Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab 28:468–481
Benarroch EE (2005) Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc 80:1326–1338
Kimelberg HK, Nedergaard M (2010) Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics 7:338–353
McGraw J, Hiebert GW, Steeves JD (2001) Modulating astrogliosis after neurotrauma. J Neurosci Res 63:109–115
Fuller S, Munch G, Steele M (2009) Activated astrocytes: a therapeutic target in Alzheimer’s disease? Expert Rev Neurother 9:1585–1594
Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14
Coco S, Calegari F, Pravettoni E et al (2003) Storage and release of ATP from astrocytes in culture. J Biol Chem 278:1354–1362
Hamilton N, Vayro S, Kirchhoff F et al (2008) Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia 56:734–749
Sawada K, Echigo N, Juge N et al (2008) Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A 105:5683–5686
Sreedharan S, Shaik JH, Olszewski PK et al (2010) Glutamate, aspartate and nucleotide transporters in the SLC17 family form four main phylogenetic clusters: evolution and tissue expression. BMC Genomics 11:17
Montana V, Malarkey EB, Verderio C et al (2006) Vesicular transmitter release from astrocytes. Glia 54:700–715
Bowser DN, Khakh BS (2007) Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol 129:485–491
Zhang Z, Chen G, Zhou W et al (2007) Regulated ATP release from astrocytes through lysosome exocytosis. Nat Cell Biol 9:945–953
Locovei S, Wang J, Dahl G (2006) Activation of pannexin 1 channels by ATP through P2Y receptors and by cytoplasmic calcium. FEBS Lett 580:239–244
Iwabuchi S, Kawahara K (2011) Functional significance of the negative-feedback regulation of ATP release via pannexin-1 hemichannels under ischemic stress in astrocytes. Neurochem Int 58:376–384
Suadicani SO, Flores CE, Urban-Maldonado M et al (2004) Gap junction channels coordinate the propagation of intercellular Ca2+ signals generated by P2Y receptor activation. Glia 48:217–229
Scemes E, Suadicani SO, Dahl G et al (2007) Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol 3:199–208
Duan S, Anderson CM, Keung EC et al (2003) P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J Neurosci 23:1320–1328
Cornell-Bell AH, Finkbeiner SM, Cooper MS et al (1990) Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247:470–473
Giaume C, Venance L (1998) Intercellular calcium signaling and gap junctional communication in astrocytes. Glia 24:50–64
Verkhratsky A, Orkand RK, Kettenmann H (1998) Glial calcium: homeostasis and signaling function. Physiol Rev 78:99–141
Newman EA, Zahs KR (1997) Calcium waves in retinal glial cells. Science 275:844–847
Guthrie PB, Knappenberger J, Segal M et al (1999) ATP released from astrocytes mediates glial calcium waves. J Neurosci 19:520–528
Cotrina ML, Lin JH, Lopez-Garcia JC et al (2000) ATP-mediated glia signaling. J Neurosci 20:2835–2844
Scemes E, Giaume C (2006) Astrocyte calcium waves: what they are and what they do. Glia 54:716–725
Araque A, Sanzgiri RP, Parpura V et al (1998) Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci 18:6822–6829
Newman EA (2003) Glial cell inhibition of neurons by release of ATP. J Neurosci 23:1659–1666
Jourdain P, Bergersen LH, Bhaukaurally K et al (2007) Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci 10:331–339
Parpura V, Haydon PG (2000) Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci U S A 97:8629–8634
Araque A, Parpura V, Sanzgiri RP et al (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215
Halassa MM, Fellin T, Haydon PG (2009) Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 57:343–346
Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32:421–431
Di Castro MA, Chuquet J, Liaudet N et al (2011) Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14:1276–1284
Panatier A, Vallee J, Haber M et al (2011) Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146:785–798
Kirischuk S, Möller T, Voitenko N et al (1995) ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosci 15:7861–7871
Franke H, Grosche J, Schädlich H et al (2001) P2X receptor expression on astrocytes in the nucleus accumbens of rats. Neuroscience 108:421–429
Kukley M, Barden JA, Steinhäuser C et al (2001) Distribution of P2X receptors on astrocytes in juvenile rat hippocampus. Glia 36:11–21
James G, Butt AM (2002) P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol 447:247–260
Fam SR, Gallagher CJ, Kalia LV et al (2003) Differential frequency dependence of P2Y1- and P2Y2- mediated Ca2+ signaling in astrocytes. J Neurosci 23:4437–4444
Gallagher CJ, Salter MW (2003) Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J Neurosci 23:6728–6739
Simard M, Arcuino G, Takano T et al (2003) Signaling at the gliovascular interface. J Neurosci 23:9254–9262
Ashour F, Deuchars J (2004) Electron microscopic localisation of P2X4 receptor subunit immunoreactivity to pre- and post-synaptic neuronal elements and glial processes in the dorsal vagal complex of the rat. Brain Res 1026:44–55
Lalo U, Pankratov Y, Wichert SP et al (2008) P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical astrocytes. J Neurosci 28:5473–5480
Fischer W, Appelt K, Grohmann M et al (2009) Increase of intracellular Ca2+ by P2X and P2Y receptor-subtypes in cultured cortical astroglia of the rat. Neuroscience 160:767–783
Nörenberg W, Schunk J, Fischer W et al (2010) Electrophysiological classification of P2X7 receptors in rat cultured neocortical astroglia. Br J Pharmacol 160:1941–1952
Oliveira JF, Riedel T, Leichsenring A et al (2011) Rodent cortical astroglia express in situ functional P2X7 receptors sensing pathologically high ATP concentrations. Cereb Cortex 21:806–820
Palygin O, Lalo U, Verkhratsky A et al (2010) Ionotropic NMDA and P2X1/5 receptors mediate synaptically induced Ca2+ signalling in cortical astrocytes. Cell Calcium 48:225–231
Franke H, Krügel U, Illes P (2006) P2 receptors and neuronal injury. Pflüger’s Arch 452:622–644
Burnstock G, Fredholm BB, Verkhratsky A (2011) Adenosine and ATP receptors in the brain. Curr Top Med Chem 11:973–1011
Burnstock G, Verkhratsky A (2010) Long-term (trophic) purinergic signalling: purinoceptors control cell proliferation, differentiation and death. Cell Death Dis 1:e9
Ballerini P, Rathbone MP, Di Iorio P et al (1996) Rat astroglial P2Z (P2X7) receptors regulate intracellular calcium and purine release. Neuroreport 7:2533–2537
Fumagalli M, Brambilla R, D’Ambrosi N et al (2003) Nucleotide-mediated calcium signaling in rat cortical astrocytes: role of P2X and P2Y receptors. Glia 43:218–230
Nobile M, Monaldi I, Alloisio S et al (2003) ATP-induced, sustained calcium signalling in cultured rat cortical astrocytes: evidence for a non-capacitative, P2X7-like-mediated calcium entry. FEBS Lett 538:71–76
Ballerini P, Giulinai P, Buccella S, Nargi E, Santavenere C, Scemes E, Rathbone MP, Caciagli F (2000) P2X7 ATP receptor mediated modulation of astrocyte proliferation and coupling. Drug Dev Res 50:108
Ciccarelli R, Ballerini P, Sabatino G et al (2001) Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci 19:395–414
Skaper SD, Debetto P, Giusti P (2010) The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J 24:337–345
Lenertz LY, Gavala ML, Zhu Y et al (2011) Transcriptional control mechanisms associated with the nucleotide receptor P2X7, a critical regulator of immunologic, osteogenic, and neurologic functions. Immunol Res 50:22–38
Jabs R, Matthias K, Grote A et al (2007) Lack of P2X receptor mediated currents in astrocytes and GluR type glial cells of the hippocampal CA1 region. Glia 55:1648–1655
Young MT, Pelegrin P, Surprenant A (2007) Amino acid residues in the P2X7 receptor that mediate differential sensitivity to ATP and BzATP. Mol Pharmacol 71:92–100
Franke H, Krügel U, Grosche J et al (2004) P2Y receptor expression on astrocytes in the nucleus accumbens of rats. Neuroscience 127:431–441
Abbracchio MP, Ceruti S (2006) Roles of P2 receptors in glial cells: focus on astrocytes. Purinergic Signal 2:595–604
Fischer W, Krügel U (2007) P2Y receptors: focus on structural, pharmacological and functional aspects in the brain. Curr Med Chem 14:2429–2455
Tonazzini I, Trincavelli ML, Montali M et al (2008) Regulation of A1 adenosine receptor functioning induced by P2Y1 purinergic receptor activation in human astroglial cells. J Neurosci Res 86:2857–2866
Tonazzini I, Trincavelli ML, Storm-Mathisen J et al (2007) Co-localization and functional cross-talk between A1 and P2Y1 purine receptors in rat hippocampus. Eur J Neurosci 26:890–902
Carrasquero LM, Delicado EG, Sanchez-Ruiloba L et al (2010) Mechanisms of protein kinase D activation in response to P2Y2 and P2X7 receptors in primary astrocytes. Glia 58:984–995
Ase AR, Bernier LP, Blais D et al (2010) Modulation of heteromeric P2X1/5 receptors by phosphoinositides in astrocytes depends on the P2X1 subunit. J Neurochem 113:1676–1684
Dusart I, Schwab ME (1994) Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci 6:712–724
Hatten ME, Liem RK, Shelanski ML et al (1991) Astroglia in CNS injury. Glia 4:233–243
Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes–implications for their role in neurologic disease. Neuroscience 54:15–36
Norenberg MD (1994) Astrocyte responses to CNS injury. J Neuropathol Exp Neurol 53:213–220
Ridet JL, Malhotra SK, Privat A et al (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20:570–577
Robson SC, Sevigny J, Zimmermann H (2006) The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal 2:409–430
Neary JT, Rathbone MP, Cattabeni F et al (1996) Trophic actions of extracellular nucleotides and nucleosides on glial and neuronal cells. Trends Neurosci 19:13–18
Abbracchio MP, Brambilla R, Ceruti S et al (1999) Signalling mechanisms involved in P2Y receptor-mediated reactive astrogliosis. Prog Brain Res 120:333–342
Neary JT, Norenberg MD (1992) Signaling by extracellular ATP: physiological and pathological considerations in neuronal–astrocytic interactions. Prog Brain Res 94:145–151
Neary JT, Whittemore SR, Zhu Q et al (1994) Destabilization of glial fibrillary acidic protein mRNA in astrocytes by ammonia and protection by extracellular ATP. J Neurochem 63:2021–2027
Neary JT, Baker L, Jorgensen SL et al (1994) Extracellular ATP induces stellation and increases glial fibrillary acidic protein content and DNA synthesis in primary astrocyte cultures. Acta Neuropathol 87:8–13
Abbracchio MP, Ceruti S, Langfelder R et al (1995) Effects of ATP analogues and basic fibroblast growth factor on astroglial cell differentiation in primary cultures of rat striatum. Int J Dev Neurosci 13:685–693
Brambilla R, Ceruti S, Malorni W et al (2000) A novel gliotic P2 receptor mediating cyclooxygenase-2 induction in rat and human astrocytes. J Auton Nerv Syst 81:3–9
Abbracchio MP, Ceruti S, Bolego C et al (1996) Trophic roles of P2 purinoceptors in central nervous system astroglial cells. CIBA Found Symp 198:142–147
Neary JT (1996) Trophic actions of extracellular ATP on astrocytes, synergistic interactions with fibroblast growth factors and underlying signal transduction mechanisms. CIBA Found Symp 198:130–139
Neary JT, McCarthy M, Cornell-Bell A et al (1999) Trophic signaling pathways activated by purinergic receptors in rat and human astroglia. Prog Brain Res 120:323–332
Rathbone MP, Middlemiss PJ, Gysbers JW et al (1999) Trophic effects of purines in neurons and glial cells. Prog Neurobiol 59:663–690
Lenz G, Goncalves D, Luo Z et al (2001) Extracellular ATP stimulates an inhibitory pathway towards growth factor-induced cRaf-1 and MEKK activation in astrocyte cultures. J Neurochem 77:1001–1009
Abbracchio MP, Saffrey MJ, Höpker V et al (1994) Modulation of astroglial cell proliferation by analogues of adenosine and ATP in primary cultures of rat striatum. Neuroscience 59:67–76
Neary JT, McCarthy M, Kang Y et al (1998) Mitogenic signaling from P1 and P2 purinergic receptors to mitogen-activated protein kinase in human fetal astrocyte cultures. Neurosci Lett 242:159–162
Franke H, Krügel U, Illes P (1999) P2 receptor-mediated proliferative effects on astrocytes in vivo. Glia 28:190–200
Franke H, Krügel U, Schmidt R et al (2001) P2 receptor-types involved in astrogliosis in vivo. Br J Pharmacol 134:1180–1189
Franke H, Sauer C, Rudolph C et al (2009) P2 receptor-mediated stimulation of the PI3-K/Akt-pathway in vivo. Glia 57:1031–1045
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Zhang D, Hu X, Qian L et al (2010) Astrogliosis in CNS pathologies: is there a role for microglia? Mol Neurobiol 41:232–241
Kuboyama K, Harada H, Tozaki-Saitoh H et al (2011) Astrocytic P2Y1 receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J Cereb Blood Flow Metab 31:1930–1941
John GR, Simpson JE, Woodroofe MN et al (2001) Extracellular nucleotides differentially regulate interleukin-1beta signaling in primary human astrocytes: implications for inflammatory gene expression. J Neurosci 21:4134–4142
Xu J, Chalimoniuk M, Shu Y et al (2003) Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, and oxidative agents. Prostaglandins Leukot Essent Fatty Acids 69:437–448
Domercq M, Brambilla L, Pilati E et al (2006) P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-α and prostaglandins. J Biol Chem 281:30684–30696
Fujita T, Tozaki-Saitoh H, Inoue K (2009) P2Y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via IL-6 release in hippocampal cultures. Glia 57:244–257
Sun JJ, Liu Y, Ye ZR (2008) Effects of P2Y1 receptor on glial fibrillary acidic protein and glial cell line-derived neurotrophic factor production of astrocytes under ischemic condition and the related signaling pathways. Neurosci Bull 24:231–243
Neary JT, Kang Y, Shi YF et al (2006) P2 receptor signalling, proliferation of astrocytes, and expression of molecules involved in cell-cell interactions. Novartis Found Symp 276:131–143
Neary JT, Kang Y (2005) Signaling from P2 nucleotide receptors to protein kinase cascades induced by CNS injury: implications for reactive gliosis and neurodegeneration. Mol Neurobiol 31:95–103
Neary JT, Shi YF, Kang Y et al (2008) Opposing effects of P2X7 and P2Y purine/pyrimidine-preferring receptors on proliferation of astrocytes induced by fibroblast growth factor-2: implications for CNS development, injury, and repair. J Neurosci Res 86:3096–3105
Tran MD, Neary JT (2006) Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proc Natl Acad Sci U S A 103:9321–9326
Tran MD, Wanner IB, Neary JT (2008) Purinergic receptor signaling regulates N-cadherin expression in primary astrocyte cultures. J Neurochem 105:272–286
Lemons ML, Howland DR, Anderson DK (1999) Chondroitin sulfate proteoglycan immunoreactivity increases following spinal cord injury and transplantation. Exp Neurol 160:51–65
Becker T, Anliker B, Becker CG et al (2000) Tenascin-R inhibits regrowth of optic fibers in vitro and persists in the optic nerve of mice after injury. Glia 29:330–346
Yang HY, Lieska N, Shao D et al (1994) Proteins of the intermediate filament cytoskeleton as markers for astrocytes and human astrocytomas. Mol Chem Neuropathol 21:155–176
Cruz-Orengo L, Figueroa JD, Velazquez I et al (2006) Blocking EphA4 upregulation after spinal cord injury results in enhanced chronic pain. Exp Neurol 202:421–433
Davalos D, Grutzendler J, Yang G et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758
Kettenmann H, Hanisch UK, Noda M et al (2011) Physiology of microglia. Physiol Rev 91:461–553
Kim JE, Kwak SE, Jo SM et al (2009) Blockade of P2X receptor prevents astroglial death in the dentate gyrus following pilocarpine-induced status epilepticus. Neurol Res 31:982–988
Inoue K, Tsuda M (2012) Purinergic systems, neuropathic pain and the role of microglia. Exp Neurol 234:293–301
Tsuda M, Tozaki-Saitoh H, Inoue K (2010) Pain and purinergic signaling. Brain Res Rev 63:222–232
Quintas C, Fraga S, Goncalves J et al (2011) P2Y receptors on astrocytes and microglia mediate opposite effects in astroglial proliferation. Purinergic Signal 7:251–263
Sun SH, Lin LB, Hung AC et al (1999) ATP-stimulated Ca2+ influx and phospholipase D activities of a rat brain-derived type-2 astrocyte cell line, RBA-2, are mediated through P2X7 receptors. J Neurochem 73:334–343
Anderson CM, Bergher JP, Swanson RA (2004) ATP-induced ATP release from astrocytes. J Neurochem 88:246–256
Wang CM, Chang YY, Kuo JS et al (2002) Activation of P2X7 receptors induced [3H]GABA release from the RBA-2 type-2 astrocyte cell line through a Cl-/HCO3 –dependent mechanism. Glia 37:8–18
Duan S, Neary JT (2006) P2X7 receptors: properties and relevance to CNS function. Glia 54:738–746
Erb L, Liao Z, Seye CI et al (2006) P2 receptors: intracellular signaling. Pflüger’s Arch 452:552–562
Bianco F, Colombo A, Saglietti L et al (2009) Different properties of P2X7 receptor in hippocampal and cortical astrocytes. Purinergic Signal 5:233–240
Adinolfi E, Cirillo M, Woltersdorf R et al (2010) Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J 24:3393–3404
Ortega F, Perez-Sen R, Delicado EG et al (2009) P2X7 nucleotide receptor is coupled to GSK-3 inhibition and neuroprotection in cerebellar granule neurons. Neurotox Res 15:193–204
Volonte C, Amadio S, D’Ambrosi N et al (2006) P2 receptor web: complexity and fine-tuning. Pharmacol Ther 112:264–280
Neary JT, Kang Y, Bu Y et al (1999) Mitogenic signaling by ATP/P2Y purinergic receptors in astrocytes: involvement of a calcium-independent protein kinase C, extracellular signal-regulated protein kinase pathway distinct from the phosphatidylinositol-specific phospholipase C/calcium pathway. J Neurosci 19:4211–4220
Lange-Carter CA, Pleiman CM, Gardner AM et al (1993) A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260:315–319
Neary JT, Zhu Q, Kang Y et al (1996) Extracellular ATP induces formation of AP-1 complexes in astrocytes via P2 purinoceptors. Neuroreport 7:2893–2896
Brambilla R, Neary JT, Cattabeni F et al (2002) Induction of COX-2 and reactive gliosis by P2Y receptors in rat cortical astrocytes is dependent on ERK1/2 but independent of calcium signalling. J Neurochem 83:1285–1296
Neary JT, Kang Y, Willoughby KA et al (2003) Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J Neurosci 23:2348–2356
Sellers LA, Simon J, Lundahl TS et al (2001) Adenosine nucleotides acting at the human P2Y1 receptor stimulate mitogen-activated protein kinases and induce apoptosis. J Biol Chem 276:16379–16390
Gendron FP, Newbold N, Vivas-Mejia P et al (2003) Signal transduction pathways for P2Y2 and P2X7 nucleotide receptors that mediate neuroinflammatory responses in astrocytes and microglial cells. Biomedical Res 14:47–61
Panenka W, Jijon H, Herx LM et al (2001) P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci 21:7135–7142
Atkins CM, Oliva AA Jr, Alonso OF et al (2007) Hypothermia treatment potentiates ERK1/2 activation after traumatic brain injury. Eur J Neurosci 26:810–819
Milenkovic I, Weick M, Wiedemann P et al (2004) Neuropeptide Y-evoked proliferation of retinal glial (Müller) cells. Graefes Arch Clin Exp Ophthalmol 242:944–950
Weisman GA, Wang M, Kong Q et al (2005) Molecular determinants of P2Y2 nucleotide receptor function: implications for proliferative and inflammatory pathways in astrocytes. Mol Neurobiol 31:169–183
Jacques-Silva MC, Rodnight R, Lenz G et al (2004) P2X7 receptors stimulate AKT phosphorylation in astrocytes. Br J Pharmacol 141:1106–1117
Burgos M, Neary JT, Gonzalez FA (2007) P2Y2 nucleotide receptors inhibit trauma-induced death of astrocytic cells. J Neurochem 103:1785–1800
Masood K, Besnard F, Su Y et al (1993) Analysis of a segment of the human glial fibrillary acidic protein gene that directs astrocyte-specific transcription. J Neurochem 61:160–166
Hope BT, Nye HE, Kelz MB et al (1994) Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron 13:1235–1244
Perez-Cadahia B, Drobic B, Davie JR (2009) H3 phosphorylation: dual role in mitosis and interphase. Biochem Cell Biol 87:695–709
Bolego C, Ceruti S, Brambilla R et al (1997) Characterization of the signalling pathways involved in ATP and basic fibroblast growth factor-induced astrogliosis. Br J Pharmacol 121:1692–1699
Tsuda M, Koizumi S, Inoue K (2001) Role of endogenous ATP at the incision area in a rat model of postoperative pain. Neuroreport 12:1701–1704
Gille H, Sharrocks AD, Shaw PE (1992) Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 358:414–417
Dash PK, Moore AN, Dixon CE (1995) Spatial memory deficits, increased phosphorylation of the transcription factor CREB, and induction of the AP-1 complex following experimental brain injury. J Neurosci 15:2030–2039
Murphy LO, Smith S, Chen RH et al (2002) Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4:556–564
Neary JT, Kang Y (2006) P2 purinergic receptors signal to glycogen synthase kinase-3beta in astrocytes. J Neurosci Res 84:515–524
Florenzano F, Viscomi MT, Amadio S et al (2008) Do ATP and NO interact in the CNS? Prog Neurobiol 84:40–56
Latini L, Geloso MC, Corvino V et al (2010) Trimethyltin intoxication up-regulates nitric oxide synthase in neurons and purinergic ionotropic receptor 2 in astrocytes in the hippocampus. J Neurosci Res 88:500–509
Washburn KB, Neary JT (2006) P2 purinergic receptors signal to STAT3 in astrocytes: Difference in STAT3 responses to P2Y and P2X receptor activation. Neuroscience 142:411–423
Peterson TS, Camden JM, Wang Y et al (2010) P2Y2 nucleotide receptor-mediated responses in brain cells. Mol Neurobiol 41:356–366
Brambilla R, Burnstock G, Bonazzi A et al (1999) Cyclo-oxygenase-2 mediates P2Y receptor-induced reactive astrogliosis. Br J Pharmacol 126:563–567
Brambilla R, Abbracchio MP (2001) Modulation of cyclooxygenase-2 and brain reactive astrogliosis by purinergic P2 receptors. Ann N Y Acad Sci 939:54–62
Phillis JW, O’Regan MH, Perkins LM (1993) Adenosine 5′-triphosphate release from the normoxic and hypoxic in vivo rat cerebral cortex. Neurosci Lett 151:94–96
Frenguelli BG, Wigmore G, Llaudet E et al (2007) Temporal and mechanistic dissociation of ATP and adenosine release during ischaemia in the mammalian hippocampus. J Neurochem 101:1400–1413
Matute C, Cavaliere F (2011) Neuroglial interactions mediated by purinergic signalling in the pathophysiology of CNS disorders. Semin Cell Dev Biol 22:252–259
Franke H, Günther A, Grosche J et al (2004) P2X7 receptor expression after ischemia in the cerebral cortex of rats. J Neuropathol Exp Neurol 63:686–699
Milius D, Gröger-Arndt H, Stanchev D et al (2007) Oxygen/glucose deprivation increases the integration of recombinant P2X7 receptors into the plasma membrane of HEK293 cells. Toxicology 238:60–69
Cavaliere F, Amadio S, Sancesario G et al (2004) Synaptic P2X7 and oxygen/glucose deprivation in organotypic hippocampal cultures. J Cereb Blood Flow Metab 24:392–398
Wirkner K, Köfalvi A, Fischer W et al (2005) Supersensitivity of P2X receptors in cerebrocortical cell cultures after in vitro ischemia. J Neurochem 95:1421–1437
Khakpay R, Polster D, Köles L et al (2010) Potentiation of the glutamatergic synaptic input to rat locus coeruleus neurons by P2X7 receptors. Purinergic Signal 6:349–359
Anderson CM, Nedergaard M (2006) Emerging challenges of assigning P2X7 receptor function and immunoreactivity in neurons. Trends Neurosci 29:257–262
Sperlagh B, Vizi ES, Wirkner K et al (2006) P2X7 receptors in the nervous system. Prog Neurobiol 78:327–346
Moran-Jimenez MJ, Matute C (2000) Immunohistochemical localization of the P2Y1 purinergic receptor in neurons and glial cells of the central nervous system. Brain Res Mol Brain Res 78:50–58
Lämmer A, Günther A, Beck A et al (2006) Neuroprotective effects of the P2 receptor antagonist PPADS on focal cerebral ischaemia-induced injury in rats. Eur J Neurosci 23:2824–2828
Lämmer AB, Beck A, Grummich B et al (2011) The P2 receptor antagonist PPADS supports recovery from experimental stroke in vivo. PLoS One 6:e19983
Darby M, Kuzmiski JB, Panenka W et al (2003) ATP released from astrocytes during swelling activates chloride channels. J Neurophysiol 89:1870–1877
Zheng W, Watts LT, Holstein DM et al (2010) Purinergic receptor stimulation reduces cytotoxic edema and brain infarcts in mouse induced by photothrombosis by energizing glial mitochondria. PLoS One 5:e14401
Wu J, Holstein JD, Upadhyay G et al (2007) Purinergic receptor-stimulated IP3-mediated Ca2+ release enhances neuroprotection by increasing astrocyte mitochondrial metabolism during aging. J Neurosci 27:6510–6520
Marrelli SP, Khorovets A, Johnson TD et al (1999) P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion. Am J Physiol 276:H33–H41
Pelligrino DA, Vetri F, Xu HL (2011) Purinergic mechanisms in gliovascular coupling. Semin Cell Dev Biol 22:229–236
Cavaliere F, Amadio S, Angelini DF et al (2004) Role of the metabotropic P2Y4 receptor during hypoglycemia: cross talk with the ionotropic NMDAR1 receptor. Exp Cell Res 300:149–158
Feeser VR, Loria RM (2011) Modulation of traumatic brain injury using progesterone and the role of glial cells on its neuroprotective actions. J Neuroimmunol 237:4–12
Hindley S, Herman MA, Rathbone MP (1994) Stimulation of reactive astrogliosis in vivo by extracellular adenosine diphosphate or an adenosine A2 receptor agonist. J Neurosci Res 38:399–406
Ryu JK, Kim J, Choi SH et al (2002) ATP-induced in vivo neurotoxicity in the rat striatum via P2 receptors. Neuroreport 13:1611–1615
Di Prospero NA, Zhou XR, Meiners S et al (1998) Suramin disrupts the gliotic response following a stab wound injury to the adult rat brain. J Neurocytol 27:491–506
Franke H, Krügel U, Grosche J, Illes P (2003) Immunoreactivity for glial fibrillary acidic protein and P2 receptor expression on astrocytes in vivo. Drug Dev Res 59:175–189
Bremicker K, Grohmann M, Dreßler J, Weber M, Franke H (2011) Human traumatic brain injury induced astrogliosis - Involvement of P2Y1 receptors? Göttingen Meeting of the German Neuroscience Society, Supplement to Neuroforum, 2011(1), Volume XVII, T9-3B (ISSN 0947–0875)
Kong Q, Peterson TS, Baker O et al (2009) Interleukin-1β enhances nucleotide-induced and alpha-secretase-dependent amyloid precursor protein processing in rat primary cortical neurons via up-regulation of the P2Y2 receptor. J Neurochem 109:1300–1310
Camden JM, Schrader AM, Camden RE et al (2005) P2Y2 nucleotide receptors enhance alpha-secretase-dependent amyloid precursor protein processing. J Biol Chem 280:18696–18702
Tran MD (2011) P2 receptor stimulation induces amyloid precursor protein production and secretion in rat cortical astrocytes. Neurosci Lett 492:155–159
Franke H (2011) Role of G protein-coupled receptors (GPCRs) for purines and pyrimidines in mediating degeneration and regeneration under neuroinflammatory processes. Purinergic Signal 7:1–5
Elliott MR, Chekeni FB, Trampont PC et al (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286
Lee M, Jantaratnotai N, McGeer E et al (2011) Mg2+ ions reduce microglial and THP-1 cell neurotoxicity by inhibiting Ca2+ entry through purinergic channels. Brain Res 1369:21–35
Delarasse C, Auger R, Gonnord P et al (2011) The purinergic receptor P2X7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J Biol Chem 286:2596–2606
Rampe D, Wang L, Ringheim GE (2004) P2X7 receptor modulation of beta-amyloid- and LPS-induced cytokine secretion from human macrophages and microglia. J Neuroimmunol 147:56–61
Sanz JM, Chiozzi P, Ferrari D et al (2009) Activation of microglia by amyloid β requires P2X7 receptor expression. J Immunol 182:4378–4385
McLarnon JG, Ryu JK, Walker DG et al (2006) Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol 65:1090–1097
Lai MK, Tan MG, Kirvell S et al (2008) Selective loss of P2Y2 nucleotide receptor immunoreactivity is associated with Alzheimer’s disease neuropathology. J Neural Transm 115:1165–1172
Moore D, Iritani S, Chambers J et al (2000) Immunohistochemical localization of the P2Y1 purinergic receptor in Alzheimer’s disease. Neuroreport 11:3799–3803
Parvathenani LK, Tertyshnikova S, Greco CR et al (2003) P2X7 mediates superoxide production in primary microglia and is up-regulated in a transgenic mouse model of Alzheimer’s disease. J Biol Chem 278:13309–13317
Mirza B, Hadberg H, Thomsen P et al (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95:425–432
Zhang YX, Yamashita H, Ohshita T et al (1995) ATP increases extracellular dopamine level through stimulation of P2Y purinoceptors in the rat striatum. Brain Res 691:205–212
Kittner H, Krügel U, Hoffmann E et al (2000) Effects of intra-accumbens injection of 2-methylthio ATP: a combined open field and electroencephalographic study in rats. Psychopharmacology (Berl) 150:123–131
Krügel U, Kittner H, Franke H et al (2001) Stimulation of P2 receptors in the ventral tegmental area enhances dopaminergic mechanisms in vivo. Neuropharmacology 40:1084–1093
Krügel U, Kittner H, Illes P (2001) Mechanisms of adenosine 5′-triphosphate-induced dopamine release in the rat nucleus accumbens in vivo. Synapse 39:222–232
Heine C, Wegner A, Grosche J et al (2007) P2 receptor expression in the dopaminergic system of the rat brain during development. Neuroscience 149:165–181
Marcellino D, Suarez-Boomgaard D, Sanchez-Reina MD et al (2010) On the role of P2X7 receptors in dopamine nerve cell degeneration in a rat model of Parkinson’s disease: studies with the P2X7 receptor antagonist A-438079. J Neural Transm 117:681–687
Schwarzschild MA (2007) Adenosine A2A antagonists as neurotherapeutics: crossing the bridge. Prog Neurobiol 83:261–262
Burnstock G, Krügel U, Abbracchio MP et al (2011) Purinergic signalling: From normal behaviour to pathological brain function. Prog Neurobiol 95:229–274
Narcisse L, Scemes E, Zhao Y et al (2005) The cytokine IL-1β transiently enhances P2X7 receptor expression and function in human astrocytes. Glia 49:245–258
Amadio S, Montilli C, Magliozzi R et al (2010) P2Y12 receptor protein in cortical gray matter lesions in multiple sclerosis. Cereb Cortex 20:1263–1273
Suadicani SO, Brosnan CF, Scemes E (2006) P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J Neurosci 26:1378–1385
Matute C, Torre I, Perez-Cerda F et al (2007) P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 27:9525–9533
Ceruti S, Vigano F, Boda E et al (2011) Expression of the new P2Y-like receptor GPR17 during oligodendrocyte precursor cell maturation regulates sensitivity to ATP-induced death. Glia 59:363–378
Fumagalli M, Daniele S, Lecca D et al (2011) Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J Biol Chem 286:10593–10604
Volonte C, Apolloni S, Carri MT et al (2011) ALS: focus on purinergic signalling. Pharmacol Ther 132:111–122
Amadio S, Apolloni S, D’Ambrosi N et al (2011) Purinergic signalling at the plasma membrane: a multipurpose and multidirectional mode to deal with amyotrophic lateral sclerosis and multiple sclerosis. J Neurochem 116:796–805
Gandelman M, Peluffo H, Beckman JS et al (2010) Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J Neuroinflammation 7:33
Jabs R, Seifert G, Steinhäuser C (2008) Astrocytic function and its alteration in the epileptic brain. Epilepsia 49(Suppl 2):3–12
Boison D (2010) Adenosine dysfunction and adenosine kinase in epileptogenesis. Open Neurosci J 4:93–101
Kumaria A, Tolias CM, Burnstock G (2008) ATP signalling in epilepsy. Purinergic Signal 4:339–346
Tian GF, Azmi H, Takano T et al (2005) An astrocytic basis of epilepsy. Nat Med 11:973–981
Etherington LA, Patterson GE, Meechan L et al (2009) Astrocytic adenosine kinase regulates basal synaptic adenosine levels and seizure activity but not activity-dependent adenosine release in the hippocampus. Neuropharmacology 56:429–437
Slezia A, Kekesi AK, Szikra T et al (2004) Uridine release during aminopyridine-induced epilepsy. Neurobiol Dis 16:490–499
Vianna EP, Ferreira AT, Naffah-Mazzacoratti MG et al (2002) Evidence that ATP participates in the pathophysiology of pilocarpine-induced temporal lobe epilepsy: fluorimetric, immunohistochemical, and Western blot studies. Epilepsia 43(Suppl 5):227–229
Avignone E, Ulmann L, Levavasseur F et al (2008) Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J Neurosci 28:9133–9144
Grosso S, Rocchi R, Margollicci M et al (2009) Postictal serum nucleotidases activities in patients with epilepsy. Epilepsy Res 84:15–20
Franke H (1995) Influence of chronic alcohol treatment on the GFAP-immunoreactivity in astrocytes of the hippocampus in rats. Acta Histochem 97:263–271
Franke H, Kittner H, Berger P et al (1997) The reaction of astrocytes and neurons in the hippocampus of adult rats during chronic ethanol treatment and correlations to behavioral impairments. Alcohol 14:445–454
Gustavsson L, Lundqvist C, Hansson E (1993) Receptor-mediated phospholipase D activity in primary astroglial cultures. Glia 8:249–255
Othman T, Sinclair CJ, Haughey N et al (2002) Ethanol alters glutamate but not adenosine uptake in rat astrocytes: evidence for protein kinase C involvement. Neurochem Res 27:289–296
Hirata H, Machado LS, Okuno CS et al (2006) Apoptotic effect of ethanol is potentiated by caffeine-induced calcium release in rat astrocytes. Neurosci Lett 393:136–140
Ruby CL, Adams CA, Knight EJ et al (2010) An essential role for adenosine signaling in alcohol abuse. Curr Drug Abuse Rev 3:163–174
Franke H, Kittner H, Grosche J et al (2003) Enhanced P2Y1 receptor expression in the brain after sensitisation with d-amphetamine. Psychopharmacology (Berl) 167:187–194
Liu JS, John GR, Sikora A et al (2000) Modulation of interleukin-1β and tumor necrosis factor α signaling by P2 purinergic receptors in human fetal astrocytes. J Neurosci 20:5292–5299
Kittner H, Grosche J, Illes P, Franke H (2003) Changes of P2Y1 receptor and iNOS-expression after sensitization with d-amphetamine by rats. Naunyn-Schmiedeberg’s Arch Pharmacol 367(1):R34–R122
Garrison CJ, Dougherty PM, Kajander KC et al (1991) Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res 565:1–7
Hald A (2009) Spinal astrogliosis in pain models: cause and effects. Cell Mol Neurobiol 29:609–619
Hald A, Nedergaard S, Hansen RR et al (2009) Differential activation of spinal cord glial cells in murine models of neuropathic and cancer pain. Eur J Pain 13:138–145
Shimizu I, Iida T, Guan Y et al (2005) Enhanced thermal avoidance in mice lacking the ATP receptor P2X3. Pain 116:96–108
Watkins LR, Milligan ED, Maier SF (2003) Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol 521:1–21
Gu JG, MacDermott AB (1997) Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 389:749–753
Werry EL, Liu GJ, Bennett MR (2006) Glutamate-stimulated ATP release from spinal cord astrocytes is potentiated by substance P. J Neurochem 99:924–936
Zhang XF, Han P, Faltynek CR et al (2005) Functional expression of P2X7 receptors in non-neuronal cells of rat dorsal root ganglia. Brain Res 1052:63–70
Zhang X, Chen Y, Wang C et al (2007) Neuronal somatic ATP release triggers neuron-satellite glial cell communication in dorsal root ganglia. Proc Natl Acad Sci U S A 104:9864–9869
Gosselin RD, Suter MR, Ji RR et al (2010) Glial cells and chronic pain. Neuroscientist 16:519–531
Baranowska-Bosiacka I, Dabrowska-Bouta B, Struzynska L (2011) Regional changes in purines and selected purinergic receptors in immature rat brain exposed to lead. Toxicology 279:100–107
Sak K, Illes P (2005) Neuronal and glial cell lines as model systems for studying P2Y receptor pharmacology. Neurochem Int 47:401–412
White N, Burnstock G (2006) P2 receptors and cancer. Trends Pharmacol Sci 27:211–217
Morrone FB, Horn AP, Stella J et al (2005) Increased resistance of glioma cell lines to extracellular ATP cytotoxicity. J Neurooncol 71:135–140
Grobben B, Claes P, Van KK et al (2001) Agonists of the P2YAC-receptor activate MAP kinase by a ras-independent pathway in rat C6 glioma. J Neurochem 78:1325–1338
Krzeminski P, Misiewicz I, Pomorski P et al (2007) Mitochondrial localization of P2Y1, P2Y2 and P2Y12 receptors in rat astrocytes and glioma C6 cells. Brain Res Bull 71:587–592
Braganhol E, Huppes D, Bernardi A et al (2009) A comparative study of ectonucleotidase and P2 receptor mRNA profiles in C6 cell line cultures and C6 ex vivo glioma model. Cell Tissue Res 335:331–340
Braganhol E, Morrone FB, Bernardi A et al (2009) Selective NTPDase2 expression modulates in vivo rat glioma growth. Cancer Sci 100:1434–1442
Morrone FB, Oliveira DL, Gamermann P et al (2006) In vivo glioblastoma growth is reduced by apyrase activity in a rat glioma model. BMC Cancer 6:226
Wink MR, Lenz G, Braganhol E et al (2003) Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett 198:211–218
Morrone FB, Jacques-Silva MC, Horn AP et al (2003) Extracellular nucleotides and nucleosides induce proliferation and increase nucleoside transport in human glioma cell lines. J Neurooncol 64:211–218
Grobben B, Claes P, Roymans D et al (2000) Ecto-nucleotide pyrophosphatase modulates the purinoceptor-mediated signal transduction and is inhibited by purinoceptor antagonists. Br J Pharmacol 130:139–145
Claes P, Grobben B, Van KK et al (2001) P2YAC–receptor agonists enhance the proliferation of rat C6 glioma cells through activation of the p42/44 mitogen-activated protein kinase. Br J Pharmacol 134:402–408
Czajkowski R, Banachewicz W, Ilnytska O et al (2004) Differential effects of P2Y1 and P2Y12 nucleotide receptors on ERK1/ERK2 and phosphatidylinositol 3-kinase signalling and cell proliferation in serum-deprived and nonstarved glioma C6 cells. Br J Pharmacol 141:497–507
White N, Ryten M, Clayton E et al (2005) P2Y purinergic receptors regulate the growth of human melanomas. Cancer Lett 224:81–91
Sabala P, Czajkowski R, Przybylek K et al (2001) Two subtypes of G protein-coupled nucleotide receptors, P2Y1 and P2Y2 are involved in calcium signalling in glioma C6 cells. Br J Pharmacol 132:393–402
Tu MT, Luo SF, Wang CC et al (2000) P2Y2 receptor-mediated proliferation of C(6) glioma cells via activation of Ras/Raf/MEK/MAPK pathway. Br J Pharmacol 129:1481–1489
Kim SG, Gao ZG, Soltysiak KA et al (2003) P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell Mol Neurobiol 23:401–418
Krzeminski P, Suplat D, Czajkowski R et al (2007) Expression and functional characterization of P2Y1 and P2Y12 nucleotide receptors in long-term serum-deprived glioma C6 cells. FEBS J 274:1970–1982
Kreda SM, Seminario-Vidal L, Heusden C et al (2008) Thrombin-promoted release of UDP-glucose from human astrocytoma cells. Br J Pharmacol 153:1528–1537
Guo LH, Trautmann K, Schluesener HJ (2004) Expression of P2X4 receptor in rat C6 glioma by tumor-associated macrophages and activated microglia. J Neuroimmunol 152:67–72
Di Virgilio F (2000) Dr. Jekyll/Mr. Hyde: the dual role of extracellular ATP. J Auton Nerv Syst 81:59–63
Neary JT, Zhu Q (1994) Signaling by ATP receptors in astrocytes. Neuroreport 5:1617–1620
James G, Butt AM (2001) Changes in P2Y and P2X purinoceptors in reactive glia following axonal degeneration in the rat optic nerve. Neurosci Lett 312:33–36
Hung AC, Chu YJ, Lin YH et al (2005) Roles of protein kinase C in regulation of P2X7 receptor-mediated calcium signalling of cultured type-2 astrocyte cell line, RBA-2. Cell Signal 17:1384–1396
Murakami K, Nakamura Y, Yoneda Y (2003) Potentiation by ATP of lipopolysaccharide-stimulated nitric oxide production in cultured astrocytes. Neuroscience 117:37–42
Kucher BM, Neary JT (2005) Bi-functional effects of ATP/P2 receptor activation on tumor necrosis factor-α release in lipopolysaccharide-stimulated astrocytes. J Neurochem 92:525–535
Gendron FP, Neary JT, Theiss PM et al (2003) Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am J Physiol Cell Physiol 284:C571–C581
Hung AC, Sun SH (2002) The P2X7 receptor-mediated phospholipase D activation is regulated by both PKC-dependent and PKC-independent pathways in a rat brain-derived type-2 astrocyte cell line, RBA-2. Cell Signal 14:83–92
Wang CM, Chang YY, Sun SH (2003) Activation of P2X7 purinoceptor-stimulated TGF-β1 mRNA expression involves PKC/MAPK signalling pathway in a rat brain-derived type-2 astrocyte cell line, RBA-2. Cell Signal 15:1129–1137
Barbieri R, Alloisio S, Ferroni S et al (2008) Differential crosstalk between P2X7 and arachidonic acid in activation of mitogen-activated protein kinases. Neurochem Int 53:255–262
King BF, Neary JT, Zhu Q et al (1996) P2 purinoceptors in rat cortical astrocytes: expression, calcium-imaging and signalling studies. Neuroscience 74:1187–1196
Centemeri C, Bolego C, Abbracchio MP et al (1997) Characterization of the Ca2+ responses evoked by ATP and other nucleotides in mammalian brain astrocytes. Br J Pharmacol 121:1700–1706
Priller J, Reddington M, Haas CA et al (1998) Stimulation of P2Y-purinoceptors on astrocytes results in immediate early gene expression and potentiation of neuropeptide action. Neuroscience 85:521–525
Fam SR, Gallagher CJ, Salter MW (2000) P2Y1 purinoceptor-mediated Ca2+ signaling and Ca2+ wave propagation in dorsal spinal cord astrocytes. J Neurosci 20:2800–2808
Chen WC, Chen CC (1998) ATP-induced arachidonic acid release in cultured astrocytes is mediated by Gi protein coupled P2Y1 and P2Y2 receptors. Glia 22:360–370
Liu J, Liao Z, Camden J et al (2004) Src homology 3 binding sites in the P2Y2 nucleotide receptor interact with Src and regulate activities of Src, proline-rich tyrosine kinase 2, and growth factor receptors. J Biol Chem 279:8212–8218
Chorna NE, Santiago-Perez LI, Erb L et al (2004) P2Y receptors activate neuroprotective mechanisms in astrocytic cells. J Neurochem 91:119–132
Wang M, Kong Q, Gonzalez FA et al (2005) P2Y nucleotide receptor interaction with alpha integrin mediates astrocyte migration. J Neurochem 95:630–640
Lenz G, Gottfried C, Luo Z et al (2000) P2Y purinoceptor subtypes recruit different Mek activators in astrocytes. Br J Pharmacol 129:927–936
Mamedova LK, Gao ZG, Jacobson KA (2006) Regulation of death and survival in astrocytes by ADP activating P2Y1 and P2Y12 receptors. Biochem Pharmacol 72:1031–1041
John GR, Scemes E, Suadicani SO et al (1999) IL-1β differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci U S A 96:11613–11618
Meme W, Ezan P, Venance L et al (2004) ATP-induced inhibition of gap junctional communication is enhanced by interleukin-1β treatment in cultured astrocytes. Neuroscience 126:95–104
Ballerini P, Di IP, Caciagli F et al (2006) P2Y2 receptor up-regulation induced by guanosine or UTP in rat brain cultured astrocytes. Int J Immunopathol Pharmacol 19:293–308
Calon F, Dridi M, Hornykiewicz O et al (2004) Increased adenosine A2A receptors in the brain of Parkinson’s disease patients with dyskinesias. Brain 127:1075–1084
Acknowledgments
The authors thank Katrin Becker, Helga Sobottka, Katrin Krause and Lutz Feige for excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (FR 1253/3-2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Franke, H., Verkhratsky, A., Burnstock, G. et al. Pathophysiology of astroglial purinergic signalling. Purinergic Signalling 8, 629–657 (2012). https://doi.org/10.1007/s11302-012-9300-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-012-9300-0