
Abstract
The neurone-centred view of the past disregarded or downplayed the role of astroglia as a primary component in the pathogenesis of neurological diseases. As this concept is changing, so is also the perceived role of astrocytes in the healthy and diseased brain and spinal cord. We have started to unravel the different signalling mechanisms that trigger specific molecular, morphological and functional changes in reactive astrocytes that are critical for repairing tissue and maintaining function in CNS pathologies, such as neurotrauma, stroke, or neurodegenerative diseases. An increasing body of evidence shows that the effects of astrogliosis on the neural tissue and its functions are not uniform or stereotypic, but vary in a context-specific manner from astrogliosis being an adaptive beneficial response under some circumstances to a maladaptive and deleterious process in another context. There is a growing support for the concept of astrocytopathies in which the disruption of normal astrocyte functions, astrodegeneration or dysfunctional/maladaptive astrogliosis are the primary cause or the main factor in neurological dysfunction and disease. This review describes the multiple roles of astrocytes in the healthy CNS, discusses the diversity of astroglial responses in neurological disorders and argues that targeting astrocytes may represent an effective therapeutic strategy for Alexander disease, neurotrauma, stroke, epilepsy and Alzheimer’s disease as well as other neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
To help and protect: the warden astrocytes
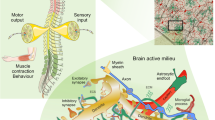
Evolution of the nervous system progressed through specialization and division of function, with neural cell networks being composed from electrically excitable neurones and electrically non-excitable glia. Neuronal specialization is in firing action potentials that propagate to axonal terminals and initiate synaptic transmission, whereas glia are optimized for housekeeping, control and neural tissue protection. The multi-partite synapse of the CNS represents a striking example of such a specialization (Fig. 1), with pre- and post-synaptic membranes being packed with exocytotic machinery, neurotransmitter receptors and proteins responsible for plasticity, whereas all “homeostatic” molecules (i.e. transporters and enzymes responsible for ion and transmitter homeostasis in the synaptic cleft, for transmitter catabolism, for metabolic support, etc.) being localized in the perisynaptic astroglial processes [235]. The synaptic assembly also includes a microglial cell process that frequently senses the synapse status [114, 229, 242]. Already at this elementary level of CNS organization, the cellular functions are divided. Neuronal compartment assures fast information flow whereas glial elements ascertain functional isolation and support of synapses, maintain synaptic operation through regulation of homeostasis and control synaptic survival or elimination depending on the network demands.

Astroglial cradle and homeostatic support of the multi-partite synapse in the CNS. The majority of synapses in the brain and in the spinal cord are multi-partite being composed of (i) the presynaptic terminal; (ii) the post-synaptic dendritic compartment; (iii) the perisynaptic process of the astrocyte; (iv) the process of neighbouring microglial cell that periodically contacts the synaptic structure and (v) the extracellular matrix (ECM) present in the synaptic cleft and also extended extra-synaptically. Astroglial perisynaptic membrane contains numerous transporters that control homeostasis in the synaptic cleft. EAAT, excitatory amino acid transporters 1 (SLC1A3) and 2 (SLC1A2); NKA, the Na+/K+ ATPase or ATP-dependent Na+/K+ pump, the α2 subtype (ATP1A2) is predominantly expressed in astrocytes; NKCC1, the Na–K-Cl co-transporter (SLC12A2); NCX, the sodium–calcium exchanger expressed in 3 isoforms (SLC8A1, SLC8A2 and SLC8A3); NAAT, the Na+-dependent ascorbic acid transporter (SLC23); NBC, the sodium-bicarbonate co-transporter (SLC4A4); CNT2, the high-affinity Na+-dependent concentrative adenosine transporter (CNT2); ASCT2, the alanine–serine–cysteine transporter 2; MCT-1, the monocarboxylase transporter 1 (SLC16A1); Kir4.1, inward rectifier Kir4.1 channels; NHE, the sodium-proton exchanger 1 (SLC9A1); GAT, GABA transporters 1 (SLC6A1) and 3 (SLC6A11); SN1,2, Na+/H+-dependent sodium-coupled neutral amino acid transporters 1 (SLC38A3) and 2 (SLC38A5); GlyT1, glycine transporter 1 (SLC6A9)
The very same specialization is observed at all levels of CNS organization. Neurones fire and establish multiple contacts whereas neuroglia control local microenvironment and protect neural tissue. In the grey matter, astrocytes divide (through the process known as tiling that starts in late embryogenesis) the parenchyma into relatively independent units traditionally known as neurovascular units, and recently often called astroglio-vascular units, that integrate, within an individual astroglial territorial domain, neural and vascular elements [33, 96, 155]. By employing a wide array of molecular mechanisms such as exocytosis, diffusion through plasmalemmal channels or membrane transporters, astrocytes secrete numerous neurotransmitters, neurohormones and trophic factors [143, 168] that regulate synaptic fields, neuronal groups and signal to other cellular elements (e.g. microglia, oligodendroglia, pericytes, endothelial cells). At the level of the whole brain, astrocytes form glia limitans, regulate emergence and function of brain–blood and brain–cerebrospinal fluid barriers and contribute to overall brain metabolism being the sole producers and repository of glycogen.
The homeostatic function of astroglia is linked to their neuroprotective capabilities, as indeed astrocytes are principal elements of CNS defence. Insults to the CNS, regardless of their aetiology, strain the organ homeostasis and these are astrocytes, which, through dedicated molecular cascades, protect neurones against glutamate excitotoxicity, extracellular K+ overload, reactive oxygen species, and these are also astrocytes that supply stressed neurones with energy substrates [222, 236]. The loss of these critical astroglial functions permits and exacerbates progression of various diseases, of which amyotrophic lateral sclerosis, toxic encephalopathies or Alzheimer's disease (AD) are prominent examples [234, 237]. Defensive function of astrocytes is manifested as reactive astrogliosis, a multicomponent and complex remodelling of astroglia triggered by lesions to the CNS [30, 169, 175, 210]. Astrogliosis is an important component of cellular pathophysiology and its suppression often aggravates neuropathology.
To learn and to remember: astroglia in adaptive and regenerative CNS plasticity
Astrocytes are fundamental elements of the adaptive plasticity of the nervous system, essential for experience-dependent learning and functional regeneration after injury. Adaptive plasticity involves dendritic and axonal arborisation, spine density, synapse number and size (structural synaptic plasticity) as well as changes in receptor composition and density, and regulation of neurotransmitter release involving individual synapses (Hebbian synaptic plasticity) or resetting the strength of all the synapses in a particular neurone (homeostatic synaptic scaling). In some brain regions such as the hippocampus and olfactory bulb, adaptive plasticity involves also changes in neuronal numbers. These structural and functional changes in neuronal networks also underlie the activity-dependent re-arrangements of cortical maps, which together with the involvement of the contra-lesional hemisphere and contra-lesional axonal remodelling contribute to the compensation and recovery of function after injury [170].
Astroglia-derived factors induce synapse formation and maturation [152]. Thrombospondin (TSP) 1 and 2, extracellular glycoproteins secreted by astrocytes, induce excitatory synapse formation during development [39] by acting through the neuronal receptor α2δ-1 [57]. Similarly, astrocytes support synaptogenesis in the regenerating post-lesioned neural tissue. Expression of TSP 1 and 2 is increased after ischemia and TSP 1- and 2-deficient mice exhibited reduced synaptic density and axonal sprouting associated with impaired motor function recovery after stroke [133]. Another thrombospondin, TSP 4, controls protective astrogenesis in the adult subventricular zone after neurotrauma [19]. Astrocyte were also reported to secrete hevin protein that stimulates synaptogenesis; conversely the protein SPARC (secreted protein acidic and rich in cysteine), also secreted by astroglia, inhibits synaptogenesis both in vitro and in vivo [120]. Astroglia-derived glypican-4 and 6 induce functional synapse maturation by increasing the number of AMPA receptors on synapses [5]. The specific function astrocytes play in regulating synapse number seems to depend on the time after injury; attenuation of reactive astrogliosis in mice led to a more prominent synaptic loss in the hippocampus in the acute phase (4 days) after injury, but led to a complete synaptic recovery by 14 days after lesion [252]. These findings indicate that while reactive astrogliosis is protective at the acute stage, if it persists, regenerative responses might be inhibited.
In the developing CNS, astrocytes acting in concert with microglia play a critical role in the elimination of supernumerary synapses and this process, called synaptic pruning, is delayed in mice in which microglial migration is suppressed due to the lack of CX3CR1 chemokine receptor [167]. Synaptic pruning in the dorsal geniculate nucleus of the thalamus is dependent on the expression of the complement protein C1q in the retinal ganglion cells [193, 221], which in turn is regulated by transforming growth factor (TGF)-β secreted by immature astrocytes in the retina [23]. The C1q activates the classical complement pathway leading to the tagging of the thalamic synapses of retinal ganglion cells with complement-derived C3b fragment; the tagged synapses are subsequently eliminated by microglia in a manner that requires the complement receptor 3, CR3 [193, 221]. In glaucoma, one of the most common neurodegenerative diseases, microglia upregulate C1q at an early stage of the disease, and mice deficient in C1q are protected against glaucoma [93]. In line with these observations, mice lacking C3, from which C3b is generated by proteolytic removal of a small peptide known as C3a, have a larger number of synapses in the hippocampal CA1 [177]. Notably, these mice do not show any spontaneous epileptiform activity, conceivably due to compensatory reduction in release probability of glutamate from the presynaptic terminals [177] but are protected from age-related hippocampal decline [202]. The complement system also contributes to axotomy-induced elimination of synapses on spinal cord motoneurones, albeit in a C1q-independent manner [20]. Remarkably, while basal levels of C3a are required for normal synaptic function, dendritic extension and neuronal maturation of neural progenitor cells [132, 204], excessive astroglial release of C3 in response to e.g. activation by β-amyloid disrupts neuronal morphology and function [132]. Astrocytes also actively engulf and eliminate synapses in the developing and adult brain. This process is governed by neuronal activity, and requires expression of phagocytic receptors MEGF10 and MERTK [40]. After ischemic injury, ephrin-A5 expressed by reactive astrocytes inhibits axonal sprouting and motor recovery [165]. Finally, astroglial cells are instrumental in controlling neuronal numbers through adult neurogenesis. Neural stem cells in the subventricular zone and in the dentate gyrus of the hippocampus, the two neurogenic regions of the adult brain, express glial fibrillary acidic protein (GFAP) and it has been proposed that many of these GFAP-positive astrocytes possess stem cell properties [28, 51, 54, 55]. Astrocytes also regulate the local microenvironment in the neurogenic regions through secreted as well as membrane-bound factors [134, 214, 251].
Astrogliopathology: classification and general concept
Fundamentally, all diseases, naturally including neurological disorders, can be broadly defined as homeostatic failures within tissue, organ or a system. For a long time neuropathology was dominated by the neurone-centric views when all conceptualisation of brain pathology was focused on neurones, on their survival or death. This neurone-centricity is now being challenged and neuroglia begin to be regarded as a central element of neuropathology [30, 175, 196, 237, 239]. The astroglial component of neuropathology is highly variable and is often disease specific. Distinct astrogliopathic changes may co-exist or emerge sequentially in the progression of neurological disorders. Here we propose a classification of astrocytopathies that is based around functional cellular response (Fig. 2). We broadly classify astrocytopathies into astroglial atrophy with loss of function, astroglial pathological remodelling and astrogliosis.

Pathological changes in astroglia
The concept of gliodegeneration in neuropathology was introduced by Emilie Croisier and Manuel Graeber in 1996 [46]. Astroglial atrophy, functionally manifested by loss of function contributes to pathological progression of a surprising variety of neurological disorders. Decrease in the astroglial numbers as well as astroglial atrophy has been detected in schizophrenia, in temporal lobe epilepsy, and in major depressive disorders and loss of astrocyte-dependent control over glutamatergic transmission is considered as one of the principal mechanisms of abnormal synaptic connectivity in these major psychiatric disorders [15, 156, 182, 238]. Astrodegeneration and downregulation of astroglial glutamate uptake plays a leading role in excitotoxicity in amyotrophic lateral sclerosis, in Korsakoff–Wernicke syndrome and in toxic encephalopathies [86, 189, 238]. Atrophic changes in astroglia are observed in several types of neurodegenerative disorders.
Pathological remodelling of astrocytes can be a causal factor in homeostatic failures of the brain such as severe leukoencephalopathy seen in Alexander disease, in which astroglial expression of mutant GFAP leads to profound deficits in the white matter [145]. Aberrant increase in the astroglial synthesis of kynurenic acid induced by infection of astrocytes with Toxoplasma gondii is considered as a risk for schizophrenia [194]. Astrogliosis represents a multifactorial and complex remodelling of astrocytes, generally characterized by an increase in expression of GFAP and vimentin as well as a profound changes in astrocytic biochemistry and physiology associated with a secretion of numerous neuroprotective and pro-inflammatory factors.
Astrocyte reactivity and reactive astrogliosis
When and where can astrocyte activation be detected and why has it evolved? Molecular and morphological features of reactive astrocytes responding to an injury or other CNS pathologies include hypertrophy of astrocyte processes and upregulation of GFAP, the key constituent of astrocyte intermediate filaments, both of these being hallmarks of reactive astrocytes in human pathologies such as neurotrauma, stroke, perinatal asphyxia, brain haemorrhage, CNS infections, epilepsy, or AD (Fig. 3). Cytokines, such as transforming growth factor (TGF)-α, ciliary neurotrophic factor (CNTF), interleukin (IL)-6, leukaemia inhibitory factor (LIF), oncostatin M all were shown to induce astrocyte activation [10, 92, 117, 181, 218, 253]. Astrocyte activation seems to be also mediated by gp-130/activator of transcription 3 (STAT3) signalling pathway, phosphorylation and nuclear translocation of STAT3, either in astrocytes themselves [218] or, indirectly, via microglia, neurones or endothelial cells. Extensive (also known as anisomorphic) reactive astrogliosis results in glial scarring, which involves not only astrocytes but also other cells types, including pericytes that were recently proposed to be key contributors to glial scars [72].

Reactive astrogliosis is the term used for responses of activated astrocytes seen in many neurological diseases. As a rule reactive astrogliosis is a defensive reaction (which is often disease-specific) in times of acute stress that aims at restoring the tissue homeostasis and restricting the damage. Persisting reactive astrogliosis has a potential of turning maladaptive, and can inhibit neural plasticity and other regenerative responses. Modified from [91, 174, 175]
Functionally, astrogliosis is aimed at: (i) increased neuroprotection and trophic support of insult-stressed neurones; (ii) isolation of the damaged area from the rest of the CNS tissue, (iii) reconstruction of the compromised blood–brain barrier; and (iv) in some situations, possibly at the facilitation of the remodelling of brain circuits in areas surrounding the lesioned region. Within the framework of neural circuit remodelling, reactive astrocytes may acquire properties of stem cells [185]. The overall result of these functional reactions is clearly beneficial for the nervous tissue, since experimental removal of reactive astrocytes increases the degree of tissue damage and neuronal death [185, 213].
Reactive astrocytes that form a border between a focal lesion and the surrounding tissue (e.g. in ischemic or traumatic lesions or around amyloid plaques in AD) provide the means to demarcate the lesion and separate it from the rest of the CNS [184, 241]. Such a sequestering of a lesion might favour clinical stabilization and allow survival, but it might negatively affect the regenerative responses at a later stage [205]. Factors reducing astrocyte activation (as measured, e.g. by the expression of GFAP) have not been sufficiently studied. Recently, complement activation-derived C3a has been shown to reduce the expression of GFAP in astrocytes subjected to ischemia while promoting their survival [203]. As the positive effect of C3a on astrocyte survival was equally strong in astrocytes lacking GFAP and vimentin, these data point to differential regulation of cell survival and GFAP expression in astrocytes in response to ischemic stress [203].
In response to injury, some reactive astrocytes proliferate and this increases the number of astrocytes at the lesion site [29, 210, 213]. Contrary to what was previously thought, recent live imaging data suggest that astrocytes do not migrate towards the side of injury [11]. Many astrocytes in the injured brain cortex become hypertrophic and up-regulate GFAP; however, they stay within their tiled domains, among which only a limited overlap can be found [11, 250]. Some astrocytes become polarized or can proliferate, with the latter ones often being associated with blood vessels [11]. Such proliferating blood vessel-associated astrocytes might regulate migration and proliferation of pericytes involved in the glial scar formation [11, 72].
Are reactive astrogliosis and corresponding changes in the astrocyte network disease specific and do they have disease-specific consequences? Recent data suggest that reactive astrogliosis has both common and unique cellular and molecular features in individual neuropathologies. Comparisons of gene expression profiles of reactive astrocytes between ischemic stroke and endotoxin-induced astrocyte activation revealed that at least half of altered gene expression was disease specific [262].
Elimination of dividing subpopulation of reactive astrocytes in transgenic mice suggested that reactive astrocytes play a positive role at the acute post-traumatic stage and limit the extent of neurodegeneration after neurotrauma [31, 60, 212]. Manipulation of reactive astrogliosis around focal lesions by ablation in astrocytes of STAT3 transcription factor, which is a transducer of signals for cytokines such as IL-6, LIF and CNTF [113, 218, 256], inhibited both astrocyte migration and lesion demarcation and resulted in larger lesions and increased functional deficit [88, 158, 247]. In contrast, ablation of Socs3, a negative feedback molecule of STAT3 [67, 148], reduced the lesion area and resulted in a better functional recovery [158].
As suggested by the above, changes associated with astrogliosis range from reversible alterations in astrocyte gene expression and cell hypertrophy with preservation of cellular domains and tissue structure, to long-lasting scar formation that involves cell proliferation and permanent rearrangement of tissue structure. Multiple lines of molecular and cellular research indicate that astrogliosis is not a simple all-or-none stereotypic program triggered by a simple on/off regulatory switch, but instead is a finely gradated continuum of changes that occur in a context-dependent manner and that can be independently regulated by a multitude of specific molecular signalling events that mediate different specific responses [210, 213]. There is growing evidence for and interest in the heterogeneity among reactive astrocytes not only across different CNS regions, but locally within the same region [7]. For example, adjacent to focal traumatic or ischemic lesions there is topographic heterogeneity of astrogliosis as regards astrocyte proliferation, morphology and gene expression with respect to distance from the insult [210, 247]. In addition, analysis at the single cell level in vivo shows that intermingled reactive astrocytes can exhibit different expression levels of (i) chemokines or cytokines [82], (ii) signalling molecules such as pSTAT3 [88], or (iii) transcription factors that regulate sonic hedgehog signalling (SHH) [69].
As outlined above, astrogliosis can be induced, regulated or modulated by a wide variety of extracellular molecules ranging from small molecules such as purines, transmitters and steroid hormones, to large polypeptide growth factors, cytokines, serum proteins or neurodegeneration-associated molecules (Fig. 4). These instructive signals can derive from many different sources and can be released by cell damage or cell death, or via specific signalling mechanisms and act via receptors that initiate intracellular second messenger signalling cascades as reviewed elsewhere [109, 172, 174, 210, 213]. Many cell types can release molecular regulators of astrogliosis, including (i) local neural and non-neural cells intrinsic to CNS tissue such as neurones, microglia, oligodendrocyte lineage cells, endothelia, pericytes, fibromeningeal cells and other astrocytes, as well as (ii) non-neural cells that gain entry into the CNS, such as bone marrow-derived leukocytes, fibrocytes, and microbial infectious agents, and (iii) cells outside of the CNS that produce serum proteins, cytokines, steroid hormones or microbial endotoxins such as lipopolysaccharides [30].

Reactive astrocytes interact with multiple cell types. a Reactive astrocytes can receive diverse molecular signals from neurons, synapses, inflammatory cells, such as microglia and white blood cells (wbc), as well as blood vessel (bv) endothelia and pericytes. Incoming molecular signals include neurone-derived growth factors and transmitters, or inflammatory cell-derived cytokines, or blood borne molecules, which then activate specific intracellular signalling pathways. b Conversely, reactive astrocytes can send diverse molecular signals that can influence all of these same cell types in context-specific manners via specific intracellular signalling pathways. Astrocyte released molecules include numerous growth factors, neurotransmitters, cytokines and chemokines. The functional implications of the diverse and complex signalling interactions of reactive astrocytes with multiple cell types are poorly understood and only beginning to be elucidated. Dissecting the functional interactions of reactive astrocytes is a next major challenge and holds much promise for improving understanding of many aspects of pathophysiology. Modified from [209]
There is now substantial information available about extra- and intracellular signalling molecules that regulate astrogliosis (Fig. 4a). Some aspects of astrogliosis can be regulated by multiple signalling cascades, while other aspects are regulated more selectively. For example, expression of intermediate filament proteins such as GFAP or vimentin can be induced by intracellular signalling pathways associated with cAMP, STAT3, NFkB, Rho-kinase, JNK, calcium and others [66, 146, 172, 174]. Similarly, astrocyte proliferation can be regulated by various extracellular signals including EGF, FGF, endothelin 1, SHH, the serum proteins thrombin and albumin and others, and by intracellular regulators such as Olig2, JNK pathway and many more [64, 127, 208, 210]. Other aspects of astrogliosis are regulated more selectively. For example, certain pro- and anti-inflammatory functions of astrocytes are regulated separately. Deletion or disruption of NFkB or SOC3 signalling pathways in astrocytes diminishes recruitment of inflammatory cells after traumatic injury and autoimmune disease [26, 27, 158]. In contrast, deletion of STAT3 or its associated membrane receptor GP130, markedly increases the spread of inflammation after traumatic injury, autoimmune disease or infection [56, 83, 88, 158, 247]. Deletion of oestrogen receptor α, but not oestrogen receptor β, selectively from astrocytes diminishes the anti-inflammatory and neuroprotective effects of oestrogen on autoimmune inflammation [216, 217]. In addition, certain microRNAs (miR) such as miR-21 and miR-181, and miR regulatory enzymes, such as Dicer, can modulate astrogliosis and its functions, adding yet another level of potential regulation and specification of functions [22, 95, 223]. Thus, different signalling mechanisms regulate different aspects of pro- or anti-inflammatory functions of reactive astrocytes.
In response to the many incoming instructive signalling events described above, reactive astrocytes can release a wide variety of instructive molecular signals that are targeted at diverse kinds of surrounding cells, including multiple types of inflammatory cells, vasculature and other non-neural cells, as well as neural cells including neurones, synapses and oligodendroglia (Fig. 4b). Substantial evidence, as revealed in particular by in vivo transgenic loss-of-function studies, indicates that via these multiple molecular signals astrogliosis exerts numerous critical functions. For example, transgenic ablation or prevention of astrogliosis or astrocyte scar formation causes increased inflammation and tissue damage and worsens functional outcome in all CNS insult models studies thus far, including traumatic injury, ischemic injury (stroke), infection, autoimmune inflammation and neurodegenerative disorders [32, 56, 60, 83, 88, 130, 142, 151, 154, 241, 247]. Nevertheless, transgenic studies also reveal the potential for certain aspects of astrogliosis to exacerbate inflammation after traumatic injury or autoimmune challenge [26, 27, 216, 217]. Large-scale gene expression evaluations also show that inflammatory mediators can drive astrocyte transcriptome profiles towards pro-inflammatory and potentially cytotoxic phenotypes [82, 262] that may be beneficial in microbial infection but may be detrimental if triggered during sterile (uninfected) tissue responses to trauma, stroke, degenerative disease or autoimmune attack [211]. Thus, transgenic loss-of-function studies point towards the potential for astrocytes to contribute to regulation of CNS inflammation in different ways, both by attracting inflammatory cells that take part in debris clearance, but also by forming scars that act as functional barriers that protect adjacent neural parenchyma from the spread of neurotoxic inflammation [30]. Together, these observations provide compelling evidence that astrogliosis exerts a variety of beneficial functions that are essential for limiting tissue damage and preserving neurological function after CNS insults but that astrogliosis also has the potential to exert detrimental effects as determined by specific signalling mechanisms.
Ablation of astrocyte intermediate filament (nanofilament) system as an experimental modulation of reactive astrogliosis
Several approaches to study the function of reactive astrocytes utilized genetic ablation of the intermediate filament proteins, the up-regulation of which represents a hallmark of reactive astrogliosis. The intermediate filament system of reactive astrocytes is composed of GFAP, vimentin and nestin, and in some astrocytes it also includes synemin [102, 176, 219]; combined deficiency of GFAP and vimentin in GFAP −/− Vim −/− mice results in a complete absence of intermediate filament in reactive astrocytes [171]. Mice with GFAP −/− Vim −/− genome show reduced reactive gliosis and glial scarring, slower healing with an increased loss of neuronal synapses following neurotrauma [171, 252], with decreased resistance of the CNS tissue to mechanical stresses [141, 232]. Astrocytes around the CNS lesion in GFAP −/− Vim −/− mice are present in normal numbers and form normally tiled domains [252], but do not develop the typical hypertrophy of the main cellular processes [250, 252]. Ischemic stroke induced in GFAP −/− Vim −/− mice results in larger infarcts [130] with the astrocyte intermediate filament system being linked to astrocyte motility [128], viscoelastic properties, which might affect cell migration [139], vesicle trafficking [179, 180, 230], activation of Erk and c-fos [153], response to hypo-osmotic and oxidative stress and neuroprotective properties [48, 53], and the efficiency of glutamate transport and astrocyte gap junctional communication [130], all of which may play roles in CNS trauma or ischemia. The causal relationship of these important associations needs to be addressed on a molecular level.
Numerous negative consequences of reactive astrogliosis were also demonstrated, in particular when it does not get resolved in time, and can thus become maladaptive [187]. The inhibition of chondroitin sulphate proteoglycans that are expressed by oligodendrocyte precursor cells and astrocytes after CNS injury is linked to improved axonal regeneration after trauma [24, 25, 45, 125, 201, 246, 260]. Ephrin-A5, expressed by reactive astrocytes after injury was shown to limit axonal sprouting and functional recovery [165]. Genetic attenuation of reactive astrogliosis in GFAP −/− Vim −/− mice also has some positive effects, albeit it is associated with more extensive tissue damage in the initial acute post-traumatic or post-ischaemic stage [130, 252]. These positive effects include improved synaptic regeneration after entorhinal cortex lesion [252], improved post-traumatic regeneration of the optic nerve in the early postnatal period [38] and improved regenerative response and functional recovery after spinal cord trauma [144]. Both basal and post-traumatic hippocampal neurogenesis are increased in GFAP −/− Vim −/− mice and it was proposed that the negative control of neurogenesis by astrocytes via Notch signalling to NSC/NPSs depends on GFAP and vimentin [251]. GFAP −/− Vim −/− mice exposed to neonatal hypoxic-ischemic injury develop normal size infarcts but show increased number of newly born cortical neurones [101]. Adult GFAP −/− Vim −/− mice support better integration of neural grafts in the retina [116] and neuronal and astrocyte differentiation of adult NSC/NPCs transplanted in the hippocampus [249]; it remains unknown whether this is caused by attenuated reactive gliosis or by altered interactions between the grafted cells and the recipient’s astrocytes. Thus, the benefits of reactive astrogliosis at the acute stress-handling phase of neurotrauma or ischemic lesions might be counterbalanced by restricted regenerative potential at a later stage.
Astroglia in neurological diseases
Genetic astrogliopathy: Alexander disease
Mutations in the astrocyte intermediate filament protein GFAP are causative for Alexander disease (AxD), a protein aggregation disorder in which the hallmark pathology consists of cytoplasmic aggregates known as Rosenthal fibres (RFs) that accumulate in the cell body, processes, and distal endfeet of astrocytes [145]. As such this disorder offers a fascinating window on the spectrum of effects that astrocyte dysfunction may have on the CNS. Clinically, patients present with a wide range of onsets from foetal through the seventh decade and varied symptomatology. Many patients exhibit some degree of white matter deficit (perhaps a combination of hypomyelination or demyelination depending on age of onset) that is typically bilaterally symmetrical and most severe in the frontal lobes but less severe or even absent in the later onset patients. A subset of patients displays focal lesions that are sometimes confused with neoplasia, especially in the brain stem. Why only certain areas of the nervous system are so vulnerable to the effects of mutations in a gene that is widely expressed, and in a cell type that is present throughout the entire nervous system, is far from clear.
One question that remains a topic of investigation is the composition of RFs and their role in disease. Early studies identified GFAP and the small heat-shock proteins aB-crystallin and Hsp27 as major components of the fibres [98]. More recently this list has expanded to include vimentin, nestin, plectin, the 20S proteasome subunit, p-JNK, p62, and synemin [146, 176, 225, 263], although the exact proportions of these various components in the fibres remain to be elucidated. A lingering question is what prompts formation of the fibres in the first place. Initial studies implicated accumulation above a critical threshold as the key, since over-expressing even wild-type GFAP to sufficient levels in mouse models leads to aggregates that are morphologically and biochemically indistinguishable (except for the absence of mutant protein) from those found in AxD [146]. What the critical threshold is remains uncertain, with data from experiments using a knock-in mouse model showing that fibres appear with a fivefold change in total brain levels [77]; experiments on similar though not identical transgenic model suggesting a much lower threshold of only 30 % excess to be sufficient [223]. In addition, no one has yet established whether RFs are protective or toxic, although evidence from the recently developed Drosophila model is compatible with the latter property [245].
In addition to the formation of RFs, a consistent downstream effect of both mutant GFAP and the accumulation of GFAP to excess is the activation of multiple stress pathways within the astrocyte. Some of these stress pathways may actually be protective, which if amplified in the proper way could be useful as therapeutic strategies. Such a role is already suggested for αB-crystallin from both the mouse and fly models [79, 245], and for the transcription factor Nrf2 from the mouse model [123]. Increased expression of GFAP itself can also be considered a type of stress response, as indicated from studies demonstrating transactivation of the Gfap gene promoter as an early event in evolution of disease [100]. Of course increasing expression of the very protein that starts the entire disease process in motion is not helpful, and more needs to be learned about how this promoter activation takes place.
Ultimately what aspects of astrocyte function are impaired via expression of mutant GFAP is still not known. Some data exists for an interference with expression of glutamate transporters, but whether this has functional significance in vivo has not yet been proven [227]. Recently, Walker et al. [243] demonstrated that the DNA- and RNA-binding protein TDP-43, clearly causative and widely implicated in other neurodegenerative disease, mis-localizes to the cytoplasm of astrocytes and becomes abnormally phosphorylated. Given the large number of genes and genetic pathways that are regulated by TDP-43, the cascade of effects initiated by GFAP mutations has the potential to quickly expand in multiple directions.
While most attention in Alexander disease research naturally has focused on astrocytes, it is worth remembering that other cells express GFAP as well, both developmentally and into adulthood. Indeed, the R236H mutant mouse suffers from a striking deficit in adult neurogenesis in the hippocampus [78]. In theory, this deficit could arise either from dysfunction of mature astrocytes in the hippocampus which are known to influence the stem cell population [12, 214], or directly from dysfunction of the stem cells themselves which also express GFAP [68, 198]. In rodents, at least, the integration of new neurones into the dentate gyrus contributes to contextual learning, spatial memory, and pattern separation [1, 50, 115, 191]. Similar claims have been made for human hippocampus [43]. The existence of this hippocampal phenotype opens an entirely new perspective and set of possibilities for studying the cognitive impairments that are frequently observed in patients with Alexander disease. Given the increasing recognition of neurogenesis as a property of the adult human central nervous system [215], it is especially valuable to have a single gene disease model in which to study the significance of adult neurogenesis.
Pathological remodelling of astroglia in epilepsy
Epilepsy is a condition of the brain characterized by the unpredictable occurrence of seizures, affecting at least 2 % of the population worldwide [90]. The vast majority of epileptic cases are of idiopathic origin with their underlying mechanisms being undefined. This disorder is generally considered to reflect neuronal malfunction, and the search for anti-epileptic drugs has largely concentrated on compounds that affect neurones. The efficacy of these drugs, old and newly created, has not improved substantially over the past decades. All known anti-epileptic drugs merely suppress symptoms without treating the underlying disorder, and at least one-third of patients are refractory to pharmacological treatment. There is, therefore, an urgent need for developing more efficacious medications. Accordingly, recent critical reviews call for alternative concepts to identify new targets for improved therapeutic approaches [65, 138, 205].
Emerging evidence suggests that astrocytes might represent such new targets. These cells are now recognized as active communication partners in the CNS. Among many homeostatic functions, astrocytes provide energetic metabolites to neurones [190], regulate K+ and glutamate homeostasis [244, 264] and synchronize neuronal firing [8, 61]. Because neurosurgical specimens from patients presenting with mesial temporal lobe epilepsy (MTLE) demonstrate marked reactive gliosis, it is conceivable that astrocytes also have a role in seizure generation and/or seizure spread. In support of this view, various membrane channels, receptors and transporters in astrocytic membranes are altered in the epileptic brain [197].
Decreased expression and function of inwardly rectifying K+ (Kir) channels characterizes astrocytes in human sclerotic hippocampus surgically resected from patients with MTLE, which indicates impaired K+ clearance and increased seizure susceptibility (reviewed by [16]; Fig. 5). Astrocytes predominantly express the Kir4.1 channel [195], and support for an anti-epileptic function of Kir4.1 came from conditional Kir4.1 knockout mice, which display an epileptic phenotype [37, 80]. Similarly, missense mutations, loss-of-function mutations or single nucleotide polymorphisms in the genes encoding Kir4.1 (and, incidentally, the water channel AQP4 usually co-localized with Kir4.1 channels in astroglial processes) are associated with human epilepsy [16].

Astrocytic dysfunction in MTLE. 1 Seizure activity leads to an increase in extracellular K+ concentration. Downregulation of Kir channels was observed in astrocytes in human and experimental epilepsy. 2 Gap junctions mediate spatial redistribution of K+ and energy metabolites. Loss of gap junction coupling in human and experimental MTLE entails impaired K+ buffering and hyperactivity. 3 Dislocation of water channels contributes to impaired K+ buffering. 4 Astrocytes accomplish glutamate uptake. Reduced expression of the astrocytic transporters (EAAT1, EAAT2) was observed in human epileptic hippocampus. Elevated extracellular glutamate decreases the threshold for seizure induction. 5 Glutamate is converted into glutamine through GS. In chronic epileptic hippocampus, loss of GS impairs extracellular glutamate clearance and glutamine supply to neurones, resulting in decreased GABA release and hyperactivity. Modified from [197]
In the adult brain, astrocytes are connected to each other through gap junctions mainly composed of connexin Cx43 and Cx30, allowing intercellular exchange of ions, amino acids and energy metabolites. This astrocytic network has important functions, including spatial buffering of K+ [244], delivery of energy metabolites to neurones [190] and regulation of adult neurogenesis [122]. In epilepsy, enhanced, reduced or unaltered expression of Cx43 and Cx30 has been reported [70, 220]. However, altered expression of connexins does not allow conclusions about functional coupling, and functional coupling studies in epilepsy are virtually absent. According to the spatial buffering concept, the astrocytic network is expected to exert anti-epileptic effects because decreased coupling would lead to accumulation of extracellular K+, neuronal depolarization and hyperactivity (Fig. 5). In accord with this idea, mice with coupling-deficient astrocytes, due to genetic deletion of Cx30 and Cx43, display impaired clearance of K+ and glutamate as well as epileptiform activity [166, 244]. However, spread of Ca2+ waves and energy supply to neurones are also reduced in the absence of astrocytic coupling, suggesting that the networks might play a dual, pro- and anti-epileptic role. These findings further emphasize the need for functional studies to unravel the role of coupling in epilepsy. Functional properties of astrocytes were recently investigated in neurosurgical hippocampal specimens from MTLE patients with and without sclerosis, combining patch clamp recording, K+ concentration analysis, EEG/video-monitoring, and fate mapping analysis [15]. The authors reported that the hippocampus of MTLE patients with sclerosis is completely devoid of bona fide astrocytes and gap junction coupling, while coupled astrocytes were abundantly present in non-sclerotic specimens (Fig. 5). To decide whether these glial changes represented cause or effect of the disease, a mouse model was established that reproduced key features of the human disease. In this model, uncoupling impaired K+ buffering and temporally preceded neuronal death and generation of spontaneous seizures. Uncoupling was induced in vivo through injection of LPS, prevented in Toll-like receptor4 knockout mice and reproduced in situ through acute IL-1β, TNFα or LPS incubation. Fate mapping confirmed that in the course of MTLE with sclerosis, astrocytes acquire an atypical functional phenotype and lose coupling [15]. The study suggested that astrocyte dysfunction might be a prime cause of the disease and identified novel targets for anti-epileptogenic therapeutic intervention.
Enhanced extracellular glutamate concentrations are observed in human epileptic tissue, which is thought to induce hyperactivity and neuronal death [71]. Whether dysfunctional glial glutamate transporters (EAAT1, EAAT2) contribute to the impaired glutamate homeostasis in epilepsy is under discussion because experimental findings are inconsistent [197]. For effective removal of excess extracellular glutamate, the transmitter must be converted by the enzyme glutamine synthetase (GS) into the receptor-inactive molecule glutamine, and recent data suggested that in epilepsy, GS might represent the bottleneck for catabolism of the transmitter [44], (Fig. 5). Indeed, loss of GS was found in the sclerotic hippocampus of MTLE patients. GS is also down-regulated in the chronic phase of experimental epilepsy, and pharmacological inhibition generated seizures and a pathology resembling human hippocampal sclerosis. Besides disturbing glutamate uptake, loss of GS also impairs delivery of glutamine to neurones by reactive astrocytes, which results in decreased GABA release from interneurones and exacerbates hyper-excitability [164], (Fig. 5).
In conclusion, although research on astrocytes in epilepsy is still in its infancy, increasing evidence suggests a critical role of these cells in the disturbance of K+ and transmitter homeostasis and seizure generation. These findings might eventually classify MTLE as a glial rather than a neuronal disorder, and identify astrocytes as promising new targets for the development of more specific anti-epileptogenic therapeutic strategies.
Astroglia in AD: reactivity, astrodegeneration and pathological remodelling
Reactivity
Astrogliosis has been reported to be an integral component of AD pathology since its first descriptions in the early twentieth century [6]. Astrocytes surrounding amyloid plaques show a reactive phenotype characterized by increased GFAP expression with hypertrophied processes which envelop and penetrate into plaques. However, the precise role of astrocyte activation in disease pathogenesis has been controversial. Activated astrocytes elaborate a complex array of inflammatory mediators. In vitro, exogenous β-amyloid stimulates astrocytes to express IL-1β, IL-6, TNF-a, IFN-γ, and iNOS [129], which have been detected in activated astrocytes surrounding plaques in transgenic mouse models and the AD brain [150, 162]. The increased expression of pro-inflammatory mediators and cytotoxic molecules in astrocytes (and other glial cells) form the basis of the “inflammation hypothesis”, which postulates that plaques activate glia and initiate a pro-inflammatory and cytotoxic cascade resulting in neurodegeneration [3].
In support of the specific role of astrocytes in mediating this effect, Furman et al. [63] demonstrated that selective inhibition of inflammatory signalling in astrocytes via viral-mediated disruption of calcineurin/NFAT (nuclear factor of activated T-cells), reduced plaque pathology and improved cognitive function in a mouse model of AD. An adeno-associated virus (AAV) driving expression of VIVIT, a peptide targeting the interaction between calcineurin and NFAT, using a GFAP promoter, was injected into the hippocampi of APP/PS1 mice. After several months, amyloid plaque load was reduced by 25 % compared to control AAV, and hippocampal-dependent active avoidance behaviour was improved. These results suggest that astrocytic inflammatory cascades regulated by calcineurin/NFAT play a critical role in exacerbating amyloid plaque pathogenesis with detrimental behavioural consequences [63].
The inflammation hypothesis gained support with early epidemiological studies which revealed that non-steroidal anti-inflammatory drug (NSAID) use was inversely correlated with the risk of AD incidence, suggesting a protective effect [140]. However, several subsequent randomized, blinded, placebo-controlled clinical trials did not confirm this beneficial effect [2, 183]. More recent studies suggest that different aspects of astrocyte function might also play a salutary role, reducing β-amyloid load during AD pathogenesis. A hallmark of astrocyte activation is the induction and assembly of the cytoplasmic intermediate filament network, consisting of GFAP and vimentin (among others), giving astrocytes the characteristic reactive phenotype [173]. Deletion of GFAP and Vim in mice results in astrocytes with peculiar phenotypes (see also above): under physiological conditions, astrocyte morphology is indistinguishable from wild-type astroglia; however, following acute CNS injury (spinal cord injury, hippocampal deafferentation, or cerebral ischemia), GFAP −/− Vim −/− astrocytes do not develop the characteristic morphologic changes associated with activation [130, 171, 252]. Similarly, gene deletion of GFAP and Vim in APP/PS1 mice resulted in alterations in the morphology of activated astrocytes. GFAP −/− Vim −/− astrocytes in close proximity to plaques had the appearance of non-reactive astrocytes with fine processes that lacked interaction with plaques, in striking contrast to the typical hypertrophied processes with intimate invasion of amyloid plaques seen with wild-type astrocytes [118]. Furthermore, the plaque load in GFAP −/− Vim −/− mice was double that found in the APP/PS1 mice with wild-type astrocytes. Of note, the finding on the amyloid plague load was not confirmed in another study with the APP/PS1 GFAP −/− Vim −/− mice, although the changes of astrocytes morphology were present [104]. In addition, GFAP and Vim absence was associated with an increased load of dystrophic neurites—the swollen neuronal processes seen adjacent to plaques—providing evidence that activated astrocytes might exert neuroprotective effects on nearby neurones. GFAP and Vim gene deletion had remarkably little effect on the expression of key cytokines and chemokines in the APP/PS1 mice: IL-1β, IL-6, IL-10, TNF-α, TGF-β, and iNOS were unchanged. The number of astrocytes, and expression of GS and S100β were also unaltered, suggesting that the absence of GFAP and vimentin had no effect on astrocyte viability. Finally, the gene deletions had little effect on APP expression or processing. Thus, the major difference between the mice seems to come from the interaction between astrocytes and amyloid plaques [118].
The precise intermediate filament-dependent mechanism by which astrocytes reduce plaque accumulation in APP/PS1 mice is not known; however, several potential mechanisms have been described. Transcriptome profiling has revealed that astrocytes express genes involved in phagocytosis, including Draper/Megf10 and Mertk/integrin αVβ5 [34]. Wyss-Coray and colleagues cultured mouse astrocytes on the surface of plaque-laden brain slices derived from aged APP transgenic mice, and found that astrocytes degraded amyloid plaques [255]. Others have demonstrated that activated astrocytes release proteases, such as matrix metalloproteinase-9, capable of degrading β-amyloid and amyloid [258, 261]. Astrocytes are capable of taking up β-amyloid, via endocytosis or macropinocytosis and subsequent trafficking and degradation via the lysosomal pathway [13, 131]. It has been hypothesized that age-dependent lysosomal dysfunction [47, 112, 254] may be an underlying mechanisms for accumulation of β-amyloid resulting from impaired degradation [62, 255]. Recent studies demonstrate that activation of ubiquitously expressed transcription factor EB (TFEB) stimulates lysosome biogenesis and cellular trafficking pathways to promote breakdown of lipids and proteins [199, 200, 257]; and remove abnormal aggregates in lysosome storage disorders [199]. The hypothesis was tested that enhancing lysosomal function in astrocytes with TFEB, would promote β-amyloid uptake and catabolism; and attenuate plaque pathogenesis. Exogenous TFEB localized to the nucleus with transcriptional induction of lysosomal biogenesis and function, in vitro. This resulted in significantly accelerated uptake of exogenously applied β-amyloid42, with increased localization to and degradation within lysosomes in primary cultures of astrocytes. Stereotactic injection of AAV particles carrying TFEB driven by a GFAP promoter was employed to achieve astrocyte-specific expression in the hippocampus of APP/PS1 transgenic mice. Viral gene transfer of TFEB to astrocyte enhanced lysosome function, resulting in reduced β-amyloid levels and shortened β-amyloid half-life in the brain interstitial fluid; and reduced amyloid plaque load in the hippocampus compared to control virus-injected mice [257]. Therefore, enhancing lysosomal function in astrocytes is an effective strategy to restore adequate β-amyloid removal and counter amyloid plaque pathogenesis in AD.
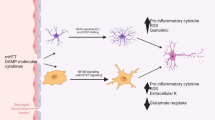
The above studies highlight the multiple facets of astrocyte activation in the setting of AD pathogenesis (Fig. 6). Astrogliosis results in the activation of a complex array of pathways involved in diverse functions, including changes in inflammation, metabolism, cytoarchitecture, and microenvironmental regulation. The activation of specific astroglial pathways (such as immune/inflammatory regulation) might exacerbate AD pathogenesis, while others (catabolic, proteolytic, and phagocytic function) might attenuate AD pathogenesis. Understanding these specific pathways as they related to disease pathogenesis will be critical for the potential identification of exploitable targets for intervention.

Multiple facets of reactive astrocytosis in Alzheimer’s disease. Schematic diagram illustrating both detrimental and salutary effects of reactive astrocytes on amyloid plaque pathogenesis. Astrocyte surrounding plaques become activated, elaborating pro-inflammatory mediators and free radicals, and may contribute to neurodegeneration. Concomitantly, astrocytosis induces catabolic, proteolytic and phagocytic pathways that might attenuate plaque pathogenesis
Astrodegeneration
Reduction in astroglial volume, surface area and in their morphological complexity has been observed in several AD transgenic mouse models [14, 121, 160, 259]. This reduction was quantified by analysing astroglial profiles labelled with antibodies against GFAP (which labels primary and possibly secondary processes) as well as with antibodies against GS and S100β (these latter stainings reveal much of astroglial arborisation including the finest processes, because both GS and S100β are cytosolic proteins whereas GFAP is associated with cytoskeleton). The total number of astrocytes labelled with these markers did not change with age in the triple transgenic model (3xTG-AD) under investigation [121, 160, 259].
The astroglial atrophy in 3xTG-AD animals was region- and age-specific with the reduction in astroglial profiles first occurred in the entorhinal cortex (at 1 month of age); in the prefrontal cortex reduction in morphological profiles became significant at 3 months of age, whereas in the hippocampus atrophic astrocytes appeared much later at 9–12 months of age (see [186, 234] for systematic review). It is of importance to observe, that atrophic astroglia emerged in all these brain regions before an appearance of extracellular β-amyloid depositions.
Morphological atrophy of astrocytes coincides with a decrease in their territorial domains, and most likely results in a reduction of the astroglial coverage of synaptic contacts belonging to these domains. Furthermore, reduced astroglial coverage may also indicate compromised homeostatic support, which may have detrimental consequences for neuroprotection and for synaptic strength and connectivity. All of this can result in decrease in the number of synapses which are known early pathological events observed in AD [41, 224]; of note a decrease in synaptic densities has been reported to correlate with the severity of dementia [49, 192]. Astrocytes support synaptic transmission through numerous coordinated mechanisms [235]; these mechanisms include regulation of ion concentrations in the synaptic cleft, shuttling lactate to active synapses, uptake of neurotransmitters and supplying neuronal terminals with glutamine, that is an obligatory precursor for glutamate and GABA. Naturally, decreased coverage of synapses by astroglial processes reduces homeostatic support and hence affects synaptic transmission.
Pathological atrophy of astroglia may also affect the neurovascular unit and reduce coverage of brain vessels with astroglial endfeet, thus contributing to vascular deficits manifest already in the early stages of AD [18, 265]. Brain metabolism is also compromised in AD and decreased glucose utilization is often detected by functional brain imaging [149]. Astrocytes are the only cells in the brain containing and processing glycogen; and astroglial metabolism was shown to be affected by β-amyloid [4]. Early stages of AD are also characterized by a remarkable decrease in noradrenergic innervations of the brain due to an early degeneration of the locus coeruleus from which noradrenergic projections originate [35]. Astroglial function, including calcium signalling, metabolism, and morphological plasticity, and gap junctional connectivity, are all controlled by noradrenergic regulation [52, 89]; the failure of the latter may further exacerbate astrodegeneration in AD.
Pathological remodelling of astrocytes: can these contribute to cognitive decline and dementia?
In the human brain, reactive astrocytes have been observed closely to Aβ plaques; however, not all Aβ plaques are surrounded by GFAP-expressing astrocytes, and reactive astrocytes also occur in areas without plaques [106, 207]. This is in contrast with an AD mouse model, in which the earliest sites of β-amyloid depositions are associated with both reactive astrocytes and microglia [105, 107]. This difference is likely due to the diversity in plaque pathology. In the human brain different plaques morphologies can be found, i.e. dense core, neuritic and diffuse plaques, some of these might be very old plaques and thus the reactive astrocyte response might have subsided. In the mouse brain a steady build up of plaques occur and the diversity of plaque morphology as in human brains is not observed. A positive correlation between GFAP expression and the neuropathological Braak stages in AD has been observed in several studies [106, 206, 248].
Astrocytes are known to be involved in clearance of Aβ [108, 255], but can they also be involved in dementia? In the rodent CNS each astrocyte supports and modulates about 100,000 synapses and this number is even higher in the human CNS where up to 2 million synapses can be supported by a single astrocyte [157]. Astrocytes in glial networks form syncytia coupled through gap junctions [70]. This property enables them to organize K+ homeostasis in the brain, an essential factor in neurone excitability (Fig. 5). Astrocytes are essential for neurotransmitter homeostasis and are actively involved in neuronal communication [81, 85]. They respond to neurotransmitters by calcium waves and release transmitters to which in turn the neurones respond [9] (Fig. 5). It has been shown that release of d-serine from astrocytes is essential for long-term potentiation in the hippocampus [87], which is a mechanism that is thought to be critical for learning and memory. Interestingly, the appearance of reactive astrocytes in the human brain, as measured by GFAP expression, coincides with the occurrence of dementia [97, 111]. This suggests that reactive astrocytes can be an important factor in the development of dementia. Furthermore, amyloid precursor protein, mutated in some forms of AD, is highly expressed in astrocytes [163], and in certain pathological conditions astrocytes can produce Aβ [231]; the Apolipoprotein E, which is the genetic risk factor for AD, is highly expressed in astrocytes [163]. A role for astrocytes in AD pathogenesis process is also supported by genome-wide association (GWAS) studies as these have revealed that many genes within GWAS loci are highly expressed in astrocytes, such as clusterin (CLU) and sortilin-related receptor L (DLR Class), A repeats containing (SORL1) [110].
The functional consequences in reactive astrocytes are yet to be fully understood. It has been shown that GS is decreased in reactive astrocytes [159] resulting in a depletion of glutamine and consequently a reduction of synaptic GABA and a hyper-excitability of hippocampal neuronal circuits [164] (Fig. 5). In an AD mouse model a hyperactivity in intracellular calcium waves in astrocytes near Aβ plaques have been observed [119]; whereas β-amyloid was shown to alter astroglial Ca2+ signalling kit [74, 188], see also [136] for detailed overview of glial calcium signalling in AD. In another AD mouse model, an increase astrocyte coupling and an increase in glutamate sensitivity was reported [178]. Furthermore, a transcriptomic profiling study on acutely isolated astrocytes showed that these cells adopt a pro-inflammatory phenotype, including up-regulation of the immunoproteasome activity [161], and a reduction in genes involved in neuronal signalling a support [162]. Recently, it was shown that reactive astrocytes in AD mice show an abundant production of and an abnormal release of the inhibitory neurotransmitter GABA, due to an increase in the enzyme MAO-B, which leads to a memory impairment in the AD mice [103].
Genetic studies have revealed causative genes and genetic risk factors, but in the majority of the AD patients the exact cause of the disease is still unclear. The disease is in about 13 % of the early-onset cases caused by autosomal recessive mutations in the genes for amyloid precursor protein (APP) and the presenilins (PSEN1 and PSEN2) [21]. ApoE is the main genetic risk factor for AD, and it has been calculated that 50 % of the late-onset AD patients have an ApoE4 allele [233]. Since 2009, more genetic risk factors have been identified with genome-wide association studies, such as clusterin, CR1, SORL1, PICALM, BIN1, EPHA1, ABCA7, MS4A, CD33 and CD2AP [21, 110]. Despite of all this knowledge, it is still elusive which molecular and cellular mechanisms cause the actual dementia. In this respect it is important to note that not only neurones are affected in AD patient brains. Astrocytes are highly involved in neuronal communication, and therefore the transformation of these cells to a reactive phenotype can have detrimental effects on the tripartite synapse. Taken together, it is imperative to consider the molecular and cellular changes of glia as well as neurones when trying to decipher the exact processes that lead to dementia in AD.
Huntington disease (HD): astroglial morpho-functional changes
In tissue of HD patients, there is a prominent astrogliosis [59, 240], which could be either primary and/or a response to neuronal dysfunction. This is characterized by a progressive increase in the number of reactive astrocytes, having hypertrophic somata and an increase in GFAP immunoreactivity and seen throughout the striatum; eventually, the blurring of the astrocytic tiling, i.e. the formation of overlapping domains between neighbouring astrocytes, occurs as the HD severity increases [59]. Similarly, astrogliosis in the striatum and cortex has been reported in many of the mouse models expressing mutated huntingtin [75, 76, 137], being more severe with animal ageing [59]. Reactive astrogliosis in HD may contribute to pericyte death, causing the reduction in pericyte coverage of cerebral blood vessels, which also could contribute to the disease progression [94].
Besides morphological changes in HD, astrocytes function is compromised as well leading to excitotoxicity, which is generally considered responsible for neuronal death [73]. There is a substantial decrease in the presence of astrocytic plasma membrane glutamate transporters EAAT2/GLT-1 along with a decrease in the astrocytic production of the antioxidant ascorbic acid [58]. The decrease in astrocytic expression of EAAT2/GLT-1 has been identified in post-mortem human tissue and in an HD mouse model [17, 59, 84, 135]. Consequently, the decreased efficacy of astrocytic glutamate uptake leads to elevated glutamate concentration in the brain, which is a leading factor in excitotoxicity and neuronal death [17, 59, 84, 135] (Fig. 5). An additional disorderly component of HD astrocytes is evident in the pathological glutamate release [126], which occurs as a result of an increased expression of the astrocyte-specific enzyme pyruvate carboxylase. As this enzyme is critical for de novo synthesis of glutamate, the resulting augmented glutamate production leads to an increased availability of cytosolic glutamate for vesicular packaging and, consequentially, pathologically high exocytotic release of this neurotransmitter from astrocytes. In addition, HD astrocytes in a different mouse model showed a decreased expression of Kir4.1 K+ channels resulting in a deficient K+ buffering (Fig. 5), which may further contribute to the pathogenesis of HD [228]. Thus, astrogliosis and dysfunctional regulation of glutamate and potassium extracellular levels by astrocytes can contribute to HD pathology. However, it remains unclear whether EAAT2, pyruvate carboxylate and Kir4.1 channels may represent targets for therapeutic interventions in HD.
Potential treatment strategies targeting astrocytes
Although much remains to be learnt about the specific involvement—primary or secondary—of astrocytes in neurological disorders, at least in some of them, astrocytes emerge as potential therapeutic targets. This was discussed above and examples of astrocyte-specific molecular targets are given in Table 1.
Conclusions
Astroglia represent the homeostatic and regulatory arm of the CNS and their dysfunction or maladaptive responses contribute to the pathogenesis of most, if not all, neurological diseases. Whether the astrocyte pathology is primary to the disease in question and how much of it is secondary is in most cases rather difficult to determine. However, even in the latter case, astrocyte dysfunction can profoundly affect and exacerbate the primary pathology. Astrocytes can contribute to neuropathology through multiple and complex pathways ranging from reactive astrogliotic response to astrodegeneration, or pathological remodelling with loss or modification of function. Astroglial reactivity is generally a defensive response aimed at containing the damage and facilitate regeneration. In certain conditions, however, pathologically modified astrocytes can release neurotoxic factors, lose intercellular communication and exacerbate vicious progression. Astrocytes, therefore, should be considered as targets for cell-specific therapy, which may open new avenues in treatment or even prevention of neurological disorders.
References
Aimone JB, Deng W, Gage FH (2011) Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70:589–596
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ et al (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289:2819–2826
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL et al (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486:410–414
Alzheimer A (1910) Beiträge zur Kenntnis der pathologischen Neuroglia und ihrer Beziehungen zu den Abbauvorgängen im Nervengewebe. In: Nissl F, Alzheimer A (eds) Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. Gustav Fischer, City, pp 401–562
Anderson MA, Ao Y, Sofroniew MV (2014) Heterogeneity of reactive astrocytes. Neurosci Lett 565C:23–29
Angulo MC, Kozlov AS, Charpak S, Audinat E (2004) Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–6927
Araque A, Carmignoto G, Haydon PG, Oliet SH, Robitaille R, Volterra A (2014) Gliotransmitters travel in time and space. Neuron 81:728–739
Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW (1994) Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci 14:846–856
Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ, Meyer-Luehmann M, Bechmann I et al (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16:580–586
Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, Gage FH, Zhao X (2006) Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Devel 15:407–421
Basak JM, Verghese PB, Yoon H, Kim J, Holtzman DM (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Abeta uptake and degradation by astrocytes. J Biol Chem 287:13959–13971
Beauquis J, Pavia P, Pomilio C, Vinuesa A, Podlutskaya N, Galvan V, Saravia F (2013) Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp Neurol 239:28–37
Bedner P, Dupper A, Huttmann K, Muller J, Herde MK, Dublin P, Deshpande T, Schramm J, Haussler U, Haas CA et al (2015) Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138:1208–1222
Bedner P, Steinhauser C (2013) Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem Int 63:682–687
Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125:1908–1922
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118:103–113
Benner EJ, Luciano D, Jo R, Abdi K, Paez-Gonzalez P, Sheng H, Warner DS, Liu C, Eroglu C, Kuo CT (2013) Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature 497:369–373
Berg A, Zelano J, Stephan A, Thams S, Barres BA, Pekny M, Pekna M, Cullheim S (2012) Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Exp Neurol 237:8–17
Bettens K, Sleegers K, Van Broeckhoven C (2013) Genetic insights in Alzheimer’s disease. Lancet Neurol 12:92–104
Bhalala OG, Pan L, Sahni V, McGuire TL, Gruner K, Tourtellotte WG, Kessler JA (2012) microRNA-21 regulates astrocytic response following spinal cord injury. J Neurosci 32:17935–17947
Bialas AR, Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16:1773–1782
Bradbury EJ, Carter LM (2011) Manipulating the glial scar: chondroitinase ABC as a therapy for spinal cord injury. Brain Res Bull 84:306–316
Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416:636–640
Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR (2005) Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 202:145–156
Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, Ivanov D, Nathanson L, Barnum SR, Bethea JR (2009) Transgenic inhibition of astroglial NF-kappaB improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol 182:2628–2640
Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M (2008) Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci 105:3581–3586
Buffo A, Rolando C, Ceruti S (2010) Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79:77–89
Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–248
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Chalermpalanupap T, Kinkead B, Hu WT, Kummer MP, Hammerschmidt T, Heneka MT, Weinshenker D, Levey AI (2013) Targeting norepinephrine in mild cognitive impairment and Alzheimer’s disease. Alzheimers Res Ther 5:21
Chen DF, Schneider GE, Martinou JC, Tonegawa S (1997) Bcl-2 promotes regeneration of severed axons in mammalian CNS. Nature 385:434–439
Chever O, Djukic B, McCarthy KD, Amzica F (2010) Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci 30:15769–15777
Cho KS, Yang L, Lu B, Feng Ma H, Huang X, Pekny M, Chen DF (2005) Re-establishing the regenerative potential of central nervous system axons in postnatal mice. J Cell Sci 118:863–872
Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433
Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504:394–400
Coleman P, Federoff H, Kurlan R (2004) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63:1155–1162
Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C (1994) Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell 79:679–694
Coras R, Siebzehnrubl FA, Pauli E, Huttner HB, Njunting M, Kobow K, Villmann C, Hahnen E, Neuhuber W, Weigel D et al (2010) Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain 133:3359–3372
Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60:1215–1226
Cregg JM, DePaul MA, Filous AR, Lang BT, Tran A, Silver J (2014) Functional regeneration beyond the glial scar. Exp Neurol 253:197–207
Croisier E, Graeber MB (2006) Glial degeneration and reactive gliosis in alpha-synucleinopathies: the emerging concept of primary gliodegeneration. Acta Neuropathol 112:517–530
Cuervo AM, Dice JF (2000) When lysosomes get old. Exp Gerontol 35:119–131
de Pablo Y, Nilsson M, Pekna M, Pekny M (2013) Intermediate filaments are important for astrocyte response to oxidative stress induced by oxygen-glucose deprivation and reperfusion. Histochem Cell Biol 140:81–91
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464
Deng W, Saxe MD, Gallina IS, Gage FH (2009) Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci 29:13532–13542
Dimou L, Gotz M (2014) Glial cells as progenitors and stem cells: new roles in the healthy and diseased brain. Physiol Rev 94:709–737
Ding F, O’Donnell J, Thrane AS, Zeppenfeld D, Kang H, Xie L, Wang F, Nedergaard M (2013) α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 54:387–394
Ding M, Eliasson C, Betsholtz C, Hamberger A, Pekny M (1998) Altered taurine release following hypotonic stress in astrocytes from mice deficient for GFAP and vimentin. Brain Res Mol Brain Res 62:77–81
Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97:703–716
Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A (1997) Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci 17:5046–5061
Drogemuller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, Deckert M, Schluter D (2008) Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol 181:2683–2693
Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD et al (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392
Estrada-Sánchez AM, Rebec GV (2012) Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: Interactions between neurons and astrocytes. Basal Ganglia 2:57–66
Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P et al (2010) In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet 19:3053–3067
Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV (2004) Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci 24:2143–2155
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743
Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, Okeda R, Ihara Y (1998) Astrocytes containing amyloid β-protein (Aβ)-positive granules are associated with Aβ40-positive diffuse plaques in the aged human brain. Am J Pathol 152:983–992
Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM (2012) Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci 32:16129–16140
Gadea A, Schinelli S, Gallo V (2008) Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci 28:2394–2408
Galanopoulou AS, Buckmaster PS, Staley KJ, Moshe SL, Perucca E, Engel J Jr, Loscher W, Noebels JL, Pitkanen A, Stables J et al (2012) Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia 53:571–582
Gao K, Wang CR, Jiang F, Wong AY, Su N, Jiang JH, Chai RC, Vatcher G, Teng J, Chen J et al (2013) Traumatic scratch injury in astrocytes triggers calcium influx to activate the JNK/c-Jun/AP-1 pathway and switch on GFAP expression. Glia 61:2063–2077
Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY (2004) Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci 101:4661–4666
Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV (2004) GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci 7:1233–1241
Garcia AD, Petrova R, Eng L, Joyner AL (2010) Sonic hedgehog regulates discrete populations of astrocytes in the adult mouse forebrain. J Neurosci 30:13597–13608
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Glass M, Dragunow M (1995) Neurochemical and morphological changes associated with human epilepsy. Brain Res Brain Res Rev 21:29–41
Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J (2011) A pericyte origin of spinal cord scar tissue. Science 333:238–242
Gray M (2014) The role of astrocytes in Huntington’s disease. In: Parpura V, Verkhratsky A (eds) Pathological potentual of neuroglia Possible new targets for medical intervention. Springer, City, pp 213–229
Grolla AA, Sim JA, Lim D, Rodriguez JJ, Genazzani AA, Verkhratsky A (2013) Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis 4:e623
Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW (2007) Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener 2:8
Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I et al (2005) Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 46:433–444
Hagemann TL, Connor JX, Messing A (2006) Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J Neurosci 26:11162–11173
Hagemann TL, Paylor R, Messing A (2013) Deficits in adult neurogenesis, contextual fear conditioning, and spatial learning in a Gfap mutant mouse model of Alexander disease. J Neurosci 33:18698–18706
Hagemann TL, Tian GF, Nedergaard M, Messing A (2008) Protective effects of αB-crystallin in mouse models of Alexander disease. Society for neurosciences abstract book
Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA (2011) Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 59:1635–1642
Halassa MM, Haydon PG (2010) Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol 72:335–355
Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV (2012) Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple g-protein-coupled receptors. J Neurosci 32:14489–14510
Haroon F, Drogemuller K, Handel U, Brunn A, Reinhold D, Nishanth G, Mueller W, Trautwein C, Ernst M, Deckert M et al (2011) Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J Immunol 186:6521–6531
Hassel B, Tessler S, Faull RL, Emson PC (2008) Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res 33:232–237
Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–1031
Hazell AS (2009) Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem Int 55:129–135
Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010) Long-term potentiation depends on release of d-serine from astrocytes. Nature 463:232–236
Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28:7231–7243
Hertz L, Chen Y, Gibbs ME, Zang P, Peng L (2004) Astrocytic adrenoceptors: a major drug target in neurological and psychiatric disorders? Curr Drug Targets CNS Neurol Disord 3:239–267
Hesdorffer DC, Logroscino G, Benn EK, Katri N, Cascino G, Hauser WA (2011) Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology 76:23–27
Hol EM, Pekny M (2015) Glial fibrillary acidic protein (GFAP) and the astrocyte intermediate filament system in diseases of the central nervous system. Curr Opin Cell Biol 32:121–130
Hostenbach S, Cambron M, D’Haeseleer M, Kooijman R, De Keyser J (2014) Astrocyte loss and astrogliosis in neuroinflammatory disorders. Neurosci Lett 565:39–41
Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M et al (2011) Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Investig 121:1429–1444
Hsiao HY, Chen YC, Huang CH, Chen CC, Hsu YH, Chen HM, Chiu FL, Kuo HC, Chang C, Chern Y (2015) Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol 78:178–192
Hutchison ER, Kawamoto EM, Taub DD, Lal A, Abdelmohsen K, Zhang Y, Wood WH 3rd, Lehrmann E, Camandola S, Becker KG et al (2013) Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia 61:1018–1028
Iadecola C, Nedergaard M (2007) Glial regulation of the cerebral microvasculature. Nat Neurosci 10:1369–1376
Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC (2004) Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62:925–931
Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE (1989) α B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell 57:71–78
Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, Shorter J (2014) Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156:170–182
Jany PL, Hagemann TL, Messing A (2013) GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro 5:e00109
Jarlestedt K, Rousset CI, Faiz M, Wilhelmsson U, Stahlberg A, Sourkova H, Pekna M, Mallard C, Hagberg H, Pekny M (2010) Attenuation of reactive gliosis does not affect infarct volume in neonatal hypoxic-ischemic brain injury in mice. PLoS One 5:e10397
Jing R, Wilhelmsson U, Goodwill W, Li L, Pan Y, Pekny M, Skalli O (2007) Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J Cell Sci 120:1267–1277
Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H et al (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896
Kamphuis W, Kooijman L, Orre M, Stassen O, Pekny M, Hol EM (2015) GFAP and vimentin deficiency alters gene expression in astrocytes and microglia in wild-type mice and changes the transcriptional response of reactive glia in mouse model for Alzheimer’s disease. Glia 63:1036–1056
Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, Verveer M, de Groot LR, Smith VD, Rangarajan S et al (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS One 7:e42823
Kamphuis W, Middeldorp J, Kooijman L, Sluijs JA, Kooi EJ, Moeton M, Freriks M, Mizee MR, Hol EM (2014) Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol Aging 35:492–510
Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM (2012) Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 60:615–629
Kanekiyo T, Xu H, Bu G (2014) ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron 81:740–754
Kang W, Hebert JM (2011) Signaling pathways in reactive astrocytes, a genetic perspective. Mol Neurobiol 43:147–154
Karch CM, Cruchaga C, Goate AM (2014) Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83:11–26
Kashon ML, Ross GW, O’Callaghan JP, Miller DB, Petrovitch H, Burchfiel CM, Sharp DS, Markesbery WR, Davis DG, Hardman J et al (2004) Associations of cortical astrogliosis with cognitive performance and dementia status. J Alzheimer’s Dis 6:595–604 (discussion 673–581)
Kato Y, Maruyama W, Naoi M, Hashizume Y, Osawa T (1998) Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. FEBS Lett 439:231–234
Kerr BJ, Patterson PH (2004) Potent pro-inflammatory actions of leukemia inhibitory factor in the spinal cord of the adult mouse. Exp Neurol 188:391–407
Kettenmann H, Kirchhoff F, Verkhratsky A (2013) Microglia: new roles for the synaptic stripper. Neuron 77:10–18
Kheirbek MA, Tannenholz L, Hen R (2012) NR2B-dependent plasticity of adult-born granule cells is necessary for context discrimination. J Neurosci 32:8696–8702
Kinouchi R, Takeda M, Yang L, Wilhelmsson U, Lundkvist A, Pekny M, Chen DF (2003) Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci 6:863–868
Klein MA, Moller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G (1997) Impaired neuroglial activation in interleukin-6 deficient mice. Glia 19:227–233
Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL et al (2013) Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J 27:187–198
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ (2009) Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–1215
Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH et al (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci 108:E440–E449
Kulijewicz-Nawrot M, Verkhratsky A, Chvatal A, Sykova E, Rodriguez JJ (2012) Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J Anat 221:252–262
Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Huttmann K, Bedner P, Requardt R, Seifert G, Redecker C, Willecke K et al (2009) Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci 106:11336–11341
LaPash Daniels CM, Austin EV, Rockney DE, Jacka EM, Hagemann TL, Johnson DA, Johnson JA, Messing A (2012) Beneficial effects of Nrf2 overexpression in a mouse model of Alexander disease. J Neurosci 32:10507–10515
Lebkuechner I, Wilhelmsson U, Mollerstrom E, Pekna M, Pekny M (2015) Heterogeneity of Notch signaling in astrocytes and the effects of GFAP and vimentin deficiency. J Neurochem 135:234–248
Lee H, McKeon RJ, Bellamkonda RV (2010) Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc Natl Acad Sci 107:3340–3345
Lee W, Reyes RC, Gottipati MK, Lewis K, Lesort M, Parpura V, Gray M (2013) Enhanced Ca2+-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol Dis 58:192–199
Levison SW, Jiang FJ, Stoltzfus OK, Ducceschi MH (2000) IL-6-type cytokines enhance epidermal growth factor-stimulated astrocyte proliferation. Glia 32:328–337
Lepekhin EA, Eliasson C, Berthold CH, Berezin V, Bock E, Pekny M (2001) Intermediate filaments regulate astrocyte motility. J Neurochem 79:617–625
Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC (2011) Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 8:67–80
Li L, Lundkvist A, Andersson D, Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Stahlberg A, Aprico K, Larsson K et al (2008) Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab 28:468–481
Li Y, Cheng D, Cheng R, Zhu X, Wan T, Liu J, Zhang R (2014) Mechanisms of U87 astrocytoma cell uptake and trafficking of monomeric versus protofibril Alzheimer’s disease amyloid-beta proteins. PLoS One 9:e99939
Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL et al (2015) NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85:101–115
Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG et al (2008) Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab 28:1722–1732
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370–1375
Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP (2001) Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis 8:807–821
Lim D, Ronco V, Grolla AA, Verkhratsky A, Genazzani AA (2014) Glial calcium signalling in Alzheimer’s disease. Rev Physiol Biochem Pharmacol 167:45–65
Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ (2001) Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet 10:137–144
Loscher W, Schmidt D (2011) Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52:657–678
Lu YB, Iandiev I, Hollborn M, Korber N, Ulbricht E, Hirrlinger PG, Pannicke T, Wei EQ, Bringmann A, Wolburg H et al (2011) Reactive glial cells: increased stiffness correlates with increased intermediate filament expression. FASEB J 25:624–631
Lucca U, Tettamanti M, Forloni G, Spagnoli A (1994) Nonsteroidal antiinflammatory drug use in Alzheimer’s disease. Biol Psychiatry 36:854–856
Lundkvist A, Reichenbach A, Betsholtz C, Carmeliet P, Wolburg H, Pekny M (2004) Under stress, the absence of intermediate filaments from Muller cells in the retina has structural and functional consequences. J Cell Sci 117:3481–3488
Macauley SL, Pekny M, Sands MS (2011) The role of attenuated astrocyte activation in infantile neuronal ceroid lipofuscinosis. J Neurosci 31:15575–15585
Malarkey EB, Parpura V (2008) Mechanisms of glutamate release from astrocytes. Neurochem Int 52:142–154
Menet V, Prieto M, Privat A, Giménez y Ribotta M (2003) Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci 100:8999–9004
Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE (2012) Alexander disease. J Neurosci 32:5017–5023
Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M (1998) Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol 152:391–398
Middeldorp J, Hol EM (2011) GFAP in health and disease. Prog Neurobiol 93:421–443
Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A (2004) Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 10:739–743
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195
Mrak RE, Griffinbc WS (2001) The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol Aging 22:915–922
Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV (2006) Essential protective roles of reactive astrocytes in traumatic brain injury. Brain 129:2761–2772
Nagler K, Mauch DH, Pfrieger FW (2001) Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol 533:665–679
Nakazawa T, Takeda M, Lewis GP, Cho KS, Jiao J, Wilhelmsson U, Fisher SK, Pekny M, Chen DF, Miller JW (2007) Attenuated glial reactions and photoreceptor degeneration after retinal detachment in mice deficient in glial fibrillary acidic protein and vimentin. Invest Ophthalmol Vis Sci 48:2760–2768
Nawashiro H, Messing A, Azzam N, Brenner M (1998) Mice lacking GFAP are hypersensitive to traumatic cerebrospinal injury. NeuroReport 9:1691–1696
Nedergaard M, Ransom B, Goldman SA (2003) New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26:523–530
Niciu MJ, Henter ID, Sanacora G, Zarate CA Jr (2014) Glial abnormalities in substance use disorders and depression: does shared glutamatergic dysfunction contribute to comorbidity? World J Biol Psychiatry 15:2–16
Oberheim NA, Wang X, Goldman S, Nedergaard M (2006) Astrocytic complexity distinguishes the human brain. Trends Neurosci 29:547–553
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y et al (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12:829–834
Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2011) Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener 6:55
Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58:831–838
Orre M, Kamphuis W, Dooves S, Kooijman L, Chan ET, Kirk CJ, Dimayuga Smith V, Koot S, Mamber C, Jansen AH et al (2013) Reactive glia show increased immunoproteasome activity in Alzheimer’s disease. Brain 136:1415–1431
Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760
Orre M, Kamphuis W, Osborn LM, Melief J, Kooijman L, Huitinga I, Klooster J, Bossers K, Hol EM (2014) Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol Aging 35:1–14
Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA (2010) Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13:584–591
Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, Dinov ID, Toga AW, Carmichael ST (2012) A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci 109:E2230–E2239
Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N (2011) Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci 108:8467–8472
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458
Parpura V, Grubisic V, Verkhratsky A (2011) Ca2+ sources for the exocytotic release of glutamate from astrocytes. Biochim Biophys Acta 1813:984–991
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF Jr, Spray DC, Reichenbach A, Pannicke T et al (2012) Glial cells in (patho)physiology. J Neurochem 121:4–27
Pekna M, Pekny M, Nilsson M (2012) Modulation of neural plasticity as a basis for stroke rehabilitation. Stroke 43:2819–2828
Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J (1999) Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol 145:503–514
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434
Pekny M, Pekna M (2004) Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol 204:428–437
Pekny M, Pekna M (2014) Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev 94:1077–1098
Pekny M, Wilhelmsson U, Pekna M (2014) The dual role of astrocyte activation and reactive gliosis. Neurosci Lett 565C:30–38
Pekny T, Faiz M, Wilhelmsson U, Curtis MA, Matej R, Skalli O, Pekny M (2014) Synemin is expressed in reactive astrocytes and Rosenthal fibers in Alexander disease. APMIS 122:76–80
Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Bjorefeldt A, Kalm M, Zetterberg H, Carlstrom KE, Blomgren K, Ekdahl CT et al (2014) Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol 253:154–164
Peters O, Schipke CG, Philipps A, Haas B, Pannasch U, Wang LP, Benedetti B, Kingston AE, Kettenmann H (2009) Astrocyte function is modified by Alzheimer’s disease-like pathology in aged mice. J Alzheimer’s Dis 18:177–189
Potokar M, Kreft M, Li L, Daniel Andersson J, Pangrsic T, Chowdhury HH, Pekny M, Zorec R (2007) Cytoskeleton and vesicle mobility in astrocytes. Traffic (Copenhagen, Denmark) 8:12–20
Potokar M, Stenovec M, Gabrijel M, Li L, Kreft M, Grilc S, Pekny M, Zorec R (2010) Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 58:1208–1219
Rabchevsky AG, Weinitz JM, Coulpier M, Fages C, Tinel M, Junier MP (1998) A role for transforming growth factor alpha as an inducer of astrogliosis. J Neurosci 18:10541–10552
Rajkowska G, Stockmeier CA (2013) Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets 14:1225–1236
Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC, Rofecoxib Protocol 091 Study G (2004) Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62:66–71
Renault-Mihara F, Okada S, Shibata S, Nakamura M, Toyama Y, Okano H (2008) Spinal cord injury: emerging beneficial role of reactive astrocytes’ migration. Int J Biochem Cell Biol 40:1649–1653
Robel S, Berninger B, Gotz M (2011) The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci 12:88–104
Rodriguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A (2015) Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience. doi:10.1016/j.neuroscience.2015.01.007
Rolls A, Shechter R, Schwartz M (2009) The bright side of the glial scar in CNS repair. Nat Rev Neurosci 10:235–241
Ronco V, Grolla AA, Glasnov TN, Canonico PL, Verkhratsky A, Genazzani AA, Lim D (2014) Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 55:219–229
Rossi D, Brambilla L, Valori CF, Roncoroni C, Crugnola A, Yokota T, Bredesen DE, Volterra A (2008) Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ 15:1691–1700
Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C (2008) Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322:1551–1555
Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R (2011) Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472:466–470
Samuel W, Masliah E, Hill LR, Butters N, Terry R (1994) Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 44:2081–2088
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74:691–705
Schwarcz R, Hunter CA (2007) Toxoplasma gondii and schizophrenia: linkage through astrocyte-derived kynurenic acid? Schizophr Bull 33:652–653
Seifert G, Huttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhauser C (2009) Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 29:7474–7488
Seifert G, Schilling K, Steinhauser C (2006) Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7:194–206
Seifert G, Steinhauser C (2013) Neuron-astrocyte signaling and epilepsy. Exp Neurol 244:4–10
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21:7153–7160
Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ et al (2013) TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15:647–658
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Sharma K, Selzer ME, Li S (2012) Scar-mediated inhibition and CSPG receptors in the CNS. Exp Neurol 237:370–378
Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC et al (2015) Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35:13029–13042
Shinjyo N, de Pablo Y, Pekny M, Pekna M (2015) Complement peptide C3a promotes astrocyte survival in response to ischemic stress. Mol Neurobiol [Epub ahead of print]
Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M (2009) Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells 27:2824–2832
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5:146–156
Simonato M, Loscher W, Cole AJ, Dudek FE, Engel J Jr, Kaminski RM, Loeb JA, Scharfman H, Staley KJ, Velisek L et al (2012) Finding a better drug for epilepsy: preclinical screening strategies and experimental trial design. Epilepsia 53:1860–1867
Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB, Function MRCC et al (2010) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31:578–590
Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans M et al (2013) Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. [corrected]. Cell Stem Cell 12:426–439
Sofroniew MV (2014) Astrogliosis. Cold Spring Harb Perspect Biol 7:a020420
Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Sofroniew MV (2014) Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 20:160–172
Sofroniew MV, Bush TG, Blumauer N, Lawrence K, Mucke L, Johnson MH (1999) Genetically-targeted and conditionally-regulated ablation of astroglial cells in the central, enteric and peripheral nervous systems in adult transgenic mice. Brain Res 835:91–95
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Song H, Stevens CF, Gage FH (2002) Astroglia induce neurogenesis from adult neural stem cells. Nature 417:39–44
Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA et al (2013) Dynamics of hippocampal neurogenesis in adult humans. Cell 153:1219–1227
Spence RD, Hamby ME, Umeda E, Itoh N, Du S, Wisdom AJ, Cao Y, Bondar G, Lam J, Ao Y et al (2011) Neuroprotection mediated through estrogen receptor-α in astrocytes. Proc Natl Acad Sci 108:8867–8872
Spence RD, Wisdom AJ, Cao Y, Hill HM, Mongerson CR, Stapornkul B, Itoh N, Sofroniew MV, Voskuhl RR (2013) Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERalpha signaling on astrocytes but not through ERbeta signaling on astrocytes or neurons. J Neurosci 33:10924–10933
Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP (2004) Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem 279:19936–19947
Stahlberg A, Andersson D, Aurelius J, Faiz M, Pekna M, Kubista M, Pekny M (2011) Defining cell populations with single-cell gene expression profiling: correlations and identification of astrocyte subpopulations. Nucleic Acids Res 39:e24
Steinhauser C, Seifert G, Bedner P (2012) Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60:1192–1202
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823
Tanaka KF, Takebayashi H, Yamazaki Y, Ono K, Naruse M, Iwasato T, Itohara S, Kato H, Ikenaka K (2007) Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia 55:617–631
Tao J, Wu H, Lin Q, Wei W, Lu XH, Cantle JP, Ao Y, Olsen RW, Yang XW, Mody I et al (2011) Deletion of astroglial dicer causes non-cell-autonomous neuronal dysfunction and degeneration. J Neurosci 31:8306–8319
Terry RD (2000) Cell death or synaptic loss in Alzheimer disease. J Neuropathol Exp Neurol 59:1118–1119
Tian R, Gregor M, Wiche G, Goldman JE (2006) Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am J Pathol 168:888–897
Tian R, Wu X, Hagemann TL, Sosunov AA, Messing A, McKhann GM, Goldman JE (2010) Alexander disease mutant glial fibrillary acidic protein compromises glutamate transport in astrocytes. J Neuropathol Exp Neurol 69:335–345
Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV et al (2014) Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci 17:694–703
Tremblay ME, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8:e1000527
Vardjan N, Gabrijel M, Potokar M, Svajger U, Kreft M, Jeras M, de Pablo Y, Faiz M, Pekny M, Zorec R (2012) IFN-γ-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J Neuroinflamm 9:144
Veeraraghavalu K, Zhang C, Zhang X, Tanzi RE, Sisodia SS (2014) Age-dependent, non-cell-autonomous deposition of amyloid from synthesis of beta-amyloid by cells other than excitatory neurons. J Neurosci 34:3668–3673
Verardo MR, Lewis GP, Takeda M, Linberg KA, Byun J, Luna G, Wilhelmsson U, Pekny M, Chen DF, Fisher SK (2008) Abnormal reactivity of muller cells after retinal detachment in mice deficient in GFAP and vimentin. Invest Ophthalmol Vis Sci 49:3659–3665
Verghese PB, Castellano JM, Holtzman DM (2011) Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol 10:241–252
Verkhratsky A, Marutle A, Rodriguez-Arellano JJ, Nordberg A (2014) Glial asthenia and functional paralysis: a new perspective on neurodegeneration and Alzheimer’s disease. Neuroscientist. pii: 1073858414547132
Verkhratsky A, Nedergaard M (2014) Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci 369:20130595
Verkhratsky A, Parpura V, Pekna M, Pekny M, Sofroniew M (2014) Glia in the pathogenesis of neurodegenerative diseases. Biochem Soc Trans 42:1291–1301
Verkhratsky A, Rodriguez JJ, Parpura V (2013) Astroglia in neurological diseases. Fut Neurol 8:149–158
Verkhratsky A, Rodriguez JJ, Steardo L (2014) Astrogliopathology: a central element of neuropsychiatric diseases? Neuroscientist 20:576–588
Verkhratsky A, Sofroniew MV, Messing A, deLanerolle NC, Rempe D, Rodriguez JJ, Nedergaard M (2012) Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro 4(3):e00082
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 44:559–577
Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980
Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A (2014) Astrocytic TDP-43 pathology in Alexander disease. J Neurosci 34:6448–6458
Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C (2006) The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26:5438–5447
Wang L, Colodner KJ, Feany MB (2011) Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J Neurosci 31:2868–2877
Wang X, Hasan O, Arzeno A, Benowitz LI, Cafferty WB, Strittmatter SM (2012) Axonal regeneration induced by blockade of glial inhibitors coupled with activation of intrinsic neuronal growth pathways. Exp Neurol 237:55–69
Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33:12870–12886
Wharton SB, O’Callaghan JP, Savva GM, Nicoll JA, Matthews F, Simpson JE, Forster G, Shaw PJ, Brayne C, Ince PG et al (2009) Population variation in glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord 27:465–473
Widestrand A, Faijerson J, Wilhelmsson U, Smith PL, Li L, Sihlbom C, Eriksson PS, Pekny M (2007) Increased neurogenesis and astrogenesis from neural progenitor cells grafted in the hippocampus of GFAP−/− Vim−/− mice. Stem Cells 25:2619–2627
Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M (2006) Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci 103:17513–17518
Wilhelmsson U, Faiz M, de Pablo Y, Sjoqvist M, Andersson D, Widestrand A, Potokar M, Stenovec M, Smith PL, Shinjyo N et al (2012) Astrocytes negatively regulate neurogenesis through the jagged1-mediated notch pathway. Stem Cells 30:2320–2329
Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE et al (2004) Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci 24:5016–5021
Winter CG, Saotome Y, Levison SW, Hirsh D (1995) A role for ciliary neurotrophic factor as an inducer of reactive gliosis, the glial response to central-nervous-system injury. Proc Natl Acad Sci 92:5865–5869
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA (2013) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 37:1949–1961
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med 9:453–457
Xia XG, Hofmann HD, Deller T, Kirsch M (2002) Induction of STAT3 signaling in activated astrocytes and sprouting septal neurons following entorhinal cortex lesion in adult rats. Mol Cell Neurosci 21:379–392
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR et al (2014) Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci 34:9607–9620
Yan P, Hu X, Song H, Yin K, Bateman RJ, Cirrito JR, Xiao Q, Hsu FF, Turk JW, Xu J et al (2006) Matrix metalloproteinase-9 degrades amyloid-beta fibrils in vitro and compact plaques in situ. J Biol Chem 281:24566–24574
Yeh CY, Vadhwana B, Verkhratsky A, Rodriguez JJ (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3:271–279
Yick LW, Wu WT, So KF, Yip HK, Shum DKY (2000) Chondroitinase ABC promotes axonal regeneration of Clarke’s neurons after spinal cord injury. NeuroReport 11:1063–1067
Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J et al (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci 26:10939–10948
Zamanian JL, Xu LJ, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410
Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 160:255–263
Zhou Y, Danbolt NC (2013) GABA and glutamate transporters in brain. Front Endocrinol 4:165
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Acknowledgments
The authors thank Roy Pekny for critical comments on the manuscript and acknowledge support from the Swedish Medical Research Council (Project 11548 and 20116), Deutsche Forschungsgemeinschaft (STE 552/3), AFA Research Foundation, ALF Göteborg (Project 11392 and 142821), Sten A. Olsson Foundation for Research and Culture, Söderberg Foundations, Hjärnfonden, Hagströmer’s Foundation Millennium, the Swedish Stroke Foundation, the Swedish Society for Medical Research, the Free Mason Foundation, Amlöv’s Foundation, E. Jacobson’s Donation Fund, NanoNet COST Action, (BM1002), the EU FP 7 Programs EduGlia (237956), NeuroGLIA (202167), EuroEPINOMICS and TargetBraIn (279017). AV was supported, in part, by the Grant (agreement from August 27 2013 No. 02.B.49.21.0003) between The Ministry of Education and Science of the Russian Federation and Lobachevsky State University of Nizhny Novgorod, by the Ministry of education of Russian Federation, unique identity number of the project is RFMEFI57814X0079, and by the grant of the Russian Scientific Foundation No. 14-15-00633. VP’s work is supported by the National Institutes of Health (HD078678). The concept of this review was conceived at the conference and training school Astrocyte Intermediate Filaments (Nanofilaments) and Astrocyte Function in Health and Disease, held at the University of Gothenburg, Sweden, in 2014, supported by NanoNet COST Action (BM1002), and the Swedish Medical Research Council, with the authors of this review as speakers.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Pekny, M., Pekna, M., Messing, A. et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol 131, 323–345 (2016). https://doi.org/10.1007/s00401-015-1513-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1513-1