
Abstract
Adenosine, a purine nucleoside, is present in all cells in tightly regulated concentrations. It has many different physiological effects in the whole body and in the heart. Adenosine activates four G protein-coupled receptors A1, A2a, A2b, and A3. Activation of myocardial A1 receptors has been shown to inhibit a variety of myocardial pathologies associated with ischemia and reperfusion injury, including stunning, arrhythmogenesis, coronary and ventricular dysfunction, acute myocardial infarction, apoptosis, and chronic heart failure, implying several options for new cardiovascular therapies for diseases, like angina pectoris, control of cardiac rhythm, ischemic injury during an acute coronary syndrome, or heart failure. However, the main issue of using full A1 receptor agonists in such indications is the broad physiologic spectrum of cardiac and extracardiac effects. Desired A1 receptor-mediated protective and regenerative cardiovascular effects might be counter-regulated by unintended side effects when considering full A1 receptor agonists. These effects can be overcome by partial A1 agonists. Partial A1 agonists can be used to trigger only some of the physiological responses of receptor activation depending on endogenous adenosine levels and on receptor reserve in different tissues. CV-Therapeutics reported the identification of a partial A1 receptor agonist CVT-3619, and recently, another partial A1 receptor agonist VCP28 was published. Both compounds are adenosine derivatives. Adenosine-like A1 receptor agonists often have the drawback of a short half-life and low bioavailability, making them not suitable for chronic oral therapy. We identified the first non-adenosine-like partial A1 receptor agonist(s) with pharmacokinetics optimal for oral once daily treatment and characterized the qualities of the partial character of the A1 receptor agonist(s) in preclinical and clinical studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adenosine, a purine nucleoside, is present in all cells in tightly regulated concentrations. It is released under a variety of physiologic and pathophysiologic conditions to facilitate protection and regeneration of injured tissues.
The adenosine-mediated effects on protection and repair can be subdivided into several categories: regulation of the oxygen supply/demand ratio to normal by reduction of oxygen demand, decrease in sympathetic tone, cell preconditioning, anti-inflammatory processes, and stimulation of angiogenesis [1, 2]. These diverse actions of adenosine are mediated by four widely expressed adenosine receptor subtypes: A1, A2a, A2b, and A3 [3].
In the heart, however, adenosine-induced protective and regenerative effects are mediated predominately by A1 receptors [4] that are expressed in cardiomyocytes [5], smooth muscle cells [5], in supraventricular tissue (atria), and ventricles [6]. A1 receptors belong to the family of seven-transmembrane-spanning purinergic P1 receptors and couple to the inhibitory Gi protein to decrease the secondary messenger cyclic adenosine monophosphate [7, 8]. Furthermore, they couple to phospholipase-C, thereby influencing inositol triphosphate and Ca2+ release from internal stores, and act on potassium and calcium channels [4].
Upon activation, myocardial A1 receptors have been shown to inhibit a variety of myocardial pathologies associated with ischemia and reperfusion injury, including stunning, arrhythmogenesis, coronary and ventricular dysfunction, acute myocardial infarction, apoptosis, and chronic heart failure [9–15], implying several options for new cardiovascular therapies for heart diseases, like angina pectoris, control of cardiac rhythm, ischemic injury during an acute coronary syndrome, or heart failure.
However, the main issue of using full A1 receptor agonists in such indications is the broad physiologic spectrum of cardiac actions, like bradycardia up to atrioventricular block (AV block), negative inotropy, and dromotropy upon adenosine A1 receptor activation [3, 4] and of extracardiac effects, such as reduction in transmitter release and locomotor activity, sedation and anticonvulsant effects in the CNS, reduction in glomerular filtration rate, inhibition of renin release, antidiuresis and vasoconstriction of afferent arteries in the kidney, and metabolism (lipolysis and glucose tolerance) [16].
Thus, the pharmacological profile of A1 receptors is manifold due to the widespread distribution and diverse function, and it should be considered that desired A1 receptor-mediated protective and regenerative cardiovascular effects might be counter-regulated by unintended side effects when considering full A1 receptor agonists. Moreover, receptor desensitization is a potential problem when considering the chronic use of full A1 receptor agonists, as it has been reported in vitro at the level of cultured atrial myocytes or in adipocytes [17, 18] and in in vivo studies [19, 20].
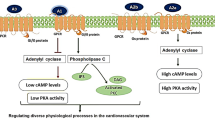
However, these restrictions for the chronic use of full A1 receptor agonists as a therapeutic option might be overcome by tailoring compounds only to the desired pharmacological efficacy by the development of partial adenosine A1 receptor agonists. A partial adenosine A1 receptor agonist is a low efficacy ligand which elicits only a submaximal response of the receptor in contrast to a full agonist (Fig. 1).

A1 receptor activation by a full or partial agonist or blockade by an antagonist in the presence of adenosine; A1R adenosine A1 receptor
Partial A1 agonists can be used to trigger only some of the physiological responses of receptor activation depending on endogenous adenosine levels and on receptor reserve in different tissues. Thus, a partial A1 receptor agonist can work as a semi-potent agonist or a weak antagonist depending on the organ/tissue adenosine A1 receptor activity leading, thereby, to a kind of tissue and functional selectivity [21]. Furthermore, partial A1 agonists might induce less receptor desensitization than full agonists [22, 23] and be ideal for chronic treatment and with broader dose ranges.
CV-Therapeutics reported the identification of a partial A1 receptor agonist CVT-3619 [24], a derivative of adenosine, which inhibits lipolysis and lowers plasma free fatty acid concentrations without any cardiovascular effects [25]. Recently, another partial A1 receptor agonist VCP28 was reported [26]. This compound, which is also an adenosine derivative, is cardioprotective at doses that have no effect on heart rate.
Adenosine-like A1 receptor agonists often have the drawback of a short half-life and low bioavailability making them not suitable for chronic oral therapy. We were aiming for non-adenosine-like partial A1 receptor agonists with pharmacokinetics optimal for oral treatment. A new class of heterocyclic compounds, devoid of the ribose moiety found in adenosine, was identified as highly potent and selective full and partial A1 agonists.
Here, we provide chemical and pharmacological data of partial A1 receptor agonists by the example of capadenoson (BAY 68-4986) (Fig. 2) [27, 28]. Capadenoson is a partial A1 receptor agonist with an EC50 of 0.1 nM on human A1 receptors and a selectivity factor of 1,800 vs. A2a, 900 vs. A2b, and no activity on A3 receptors. In consistency with the predicted pharmacological effects of a partial A1 receptor agonist, capadenoson showed reduced bradycardiac effects in preclinical models and no effects on heart rate at rest in clinical studies. The full cardioprotective potential of the compound could be demonstrated in preclinical models of acute myocardial infarction. Capadenoson is, therefore, an example of a partial A1 agonist with a highly sophisticated pharmacological activity, providing further evidence that partial agonists can be tailored toward desired pharmacological effects by reducing undesired ones.

Chemical structure of capadenoson
Material and methods
Optimization of partial adenosine A1 receptor agonists
Optimization of partial A1 agonists has been described in great detail by Nell and Albrecht-Küpper [28].
35S-GTP binding assay
Human frontal lobe was obtained from Analytical Biological Services Inc. (Wilmington, DE, USA). Membranes of the cortex were prepared as described previously [29]. [35S]GTPγS binding was measured as described by Lorenzen et al. [30].
Briefly, 5 μg of membrane protein was incubated with capadenoson or 2-chloro-N6-cyclopentyladenosine (CCPA), each in a concentration range of 0.1 nM–10 μM, in 160 μl binding buffer 50 mM Tris/HCl, pH 7.4, 2 mM triethylamine, 1 mM EDTA, 5 mM MgCl2, 10 μM GDP, 1 mM dithiothreitol, 100 mM NaCl 0.2 U/ml adenosine deaminase, 0.2 nM [35S]GTPγS, and 0.5% bovine serum albumin for 2 h at 25°C. Non-specific binding was determined in the presence of 10 μM GTPγS. Binding was determined after filtration over multiscreen FB fiber glass filters and scintillation measurement. Binding curves of [35S]GTPγS were analyzed by nonlinear regression.
Langendorff hearts
Male Wistar rats (200–250 g) were anesthetized using pentobarbital (100 mg/kg i.p.). The heart was rapidly excised and connected to a Langendorff perfusion system (FMI GmbH, Seeheim-Ober Beerbach, Germany). The heart was perfused at a constant rate of 10 ml/min with Krebs–Henseleit bicarbonate buffer solution equilibrated with 95% O2–5% CO2. The perfusion solution contained (in millimolars): NaCl 118, KCl 3, NaHCO3 22, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.8, Glucose 10, and Na-Pyruvat 2. A pressure transducer registered the perfusion pressure in the perfusion system and heart rate. The hearts were spontaneously beating. The signals from the pressure transducer were amplified, registered, and used for the calculation of the heart frequency by a personal computer. Compounds (BAY 68-4986, CCPA) were given as stock solutions directly to the perfusion buffer to result in final increasing concentrations of 1 nM–1 μM on eight to ten hearts.
Acute myocardial infarction
Male Wistar rats (Harlan-Winkelmann, 280–300 g) were treated with aspirin (2 mg/kg; p.o.) 1 h before surgery. Rats were anesthetized with pentobarbital (80 mg/kg; i.p.) and ventilated. The chest was opened and a thread was put around the LAD. Fifteen minutes before occlusion placebo, 0.03, 0.1, and 0.3 mg/kg of capadenoson were given intravenously as bolus injection. The LAD was occluded for 30 min and then re-opened. Reperfusion was allowed for 120 min. Animals were sacrificed and hearts removed. The LAD was occluded again and hearts were retrograde perfused with 0.1% Evans blue for 10 min. Hearts were cut in 2-mm slices and stained in 1% triphenyl tetrazolium chloride for 20 min at 37°C. Infarct sizes (IS) were determined by computer planimetry of the heart slices as % IS of area at risk. Statistical analysis was done by one-way ANOVA.
Results
Capadenoson
Capadenoson was identified as a result of screening on a high-throughput scale together with a thorough lead optimization program. During the course of the project, several high-throughput screening of CHO cell lines expressing human adenosine receptors and luciferase or aequorin reporter genes revealed the lead compound (2, Fig. 3) derived from one of the hit clusters. The lead compound already exhibited high affinity for the A1 receptor; however, selectivity against the A2b receptor was rather low. Under further evaluation, the compound revealed liabilities that included a marked hepatic metabolism and insufficient pharmacokinetic parameters for p.o. application. Addressing the poor selectivity for the A1 receptor, compound 3 (Table 1) was synthesized. Introduction of the hydroxyethyl residue led to a selectivity of approximately 100 over the A2b receptor and of more than 3,000 over the A2a and A3 receptors. Based on this compound, a structure activity relationship was established. The phenolic group at the northern site offered an easy point of variation (Table 2). Several residues were tested including the replacement of the oxygen atom by other heteroatoms. The hydroxyalkyl derivatives proved to be the most potent and selective compounds within this series. Several substitutions were introduced at the southeastern site to explore the influence on potency and selectivity (Table 3). Compounds bearing [2-aryl-1,3-thiazol-4-yl]methyl and [2-morpholinyl-1,3-thiazol-4-yl]methyl groups resulted in the most favorable profile. Substitution at the amino group in the southwest region was explored to some extent; however, the amino group proved to be the group of choice (Table 4). Core variations, e.g., replacement of the dicyanopyridine by cyanopyrimidine resulted in less active compounds, with some compounds exhibiting antagonistic activity for the adenosine receptors.

Chemical synthesis of a none-purinergic adenosine A1 agonist
Further optimization of various parameters based on the established SAR led to the final candidate, capadenoson. The synthesis of capadenoson [31] is straight forward starting from the aromatic aldehyde which is reacted in a Knoevenagel reaction with dicyanomethane and subsequently treated with cyanothioacetamide and the corresponding alkylchloro building block to form capadenoson via the thiol-substituted dicyanopyridine ring system. The required alkylchloro building block is formed by reaction of p-chloroarylthioacetamide with 1,3-dichloroacetone (Fig. 4).

Chemical synthesis of capadenoson
Partial character of capadenoson
The partial character of capadenoson has been evaluated by [35S]-GTPγS binding assay on preparations of human frontal lobe membranes. CCPA, a well-described full A1 receptor agonist, with a comparable potency (EC50 0.3 nM) to capadenoson (EC50 0.1 nM) on the human A1 receptor, was used as control for maximal binding of [35S]-GTPγS binding. As shown in Fig. 5, capadenoson reached only 74 ± 2% of the full effect of CCPA demonstrating the partial character of the compound.

Stimulation (in percent) of [35S]GTPγS binding by capadenoson (circle) or CCPA (triangle) to human cortex membranes. Maximal CCPA stimulation is set as 100%. Shown are means ± SEM
Bradycardia in Langendorff hearts
The full adenosine A1 receptor agonist CCPA reduced heart rate significantly in isolated rat Langendorff hearts in a concentration-dependent manner. Starting from 320 ± 2 bpm, heart rate was reduced down to 80 ± 5 bpm in a dose range from 1 nM to 1 μM (Fig. 6). Higher concentrations resulted in a complete AV block. In contrast, capadenoson reduced heart rate only from 360 ± 3 to 330 ± 6 beats/min in the same concentration range. No AV block could be induced even at concentrations of capadenoson up to 10 μM.

Reduction of heart rate by increasing concentrations of capadenoson (black bars) and CCPA (white bars) in isolated rat Langendorff hearts. Shown are means ± SEM
Cardioprotection
A model of acute myocardial infarction was used to demonstrate cardioprotective effects of capadenoson. Transient occlusion of the LAD resulted in the induction of myocardial infarction. Placebo animals reached an IS of 29 ± 2% of the area at risk (Fig. 7). Pre-ischemic treatment of rats with capadenoson decreased dose-dependently IS significantly by 30% down to 21 ± 3% at 0.3 mg/kg.

Dose-dependent reduction of infarct size by capadenoson in an ischemia–reperfusion injury rat model. Capadenoson was given in mg/kg i.v. Shown are means ± SEM. n = 9–18. *p < 0.05; **p < 0.01 vs. placebo
Discussion
There are several options for new therapies involving A1 receptor activation in cardiovascular diseases such as angina pectoris, control of cardiac rhythm, and ischemic injury during an acute coronary syndrome or coronary artery bypass graft surgery and heart failure. But besides the predicted benefits of A1 receptor activation, there are several pharmacological effects which would render a full A1 agonist to be not suitable for treatment. For example, an A1 receptor agonist which is used for cardioprotection in patients with cardiovascular diseases should not impair renal function.
This differentiation of pharmacological activities needed for therapeutic use can be achieved by the development of partial A1 receptor agonists. During the optimization of non-adenosine-like A1 receptor agonists, we noticed that the degree of partiality on the A1 receptor determines its pharmacological profile. Reduction of heart rate by activation of A1 receptors in the AV node, thus the slowing of conduction and the transfer time of signaling from atria to ventricles, is highly dependent on complete adenosine A1 receptor activation. Preclinical and clinical studies with full A1 receptor agonists, such as tecadenoson, an N6-substituted derivative of adenosine, and selodenoson [27], showed significant reduction of heart rate up to a third degree AV block in animal models [32] and in patients [33].
In contrast, the partial A1 receptor agonist capadenoson reduced heart rate only slightly in animal models, as shown by our results and had no significant effect on heart rate at rest in healthy subjects or patients with atrial fibrillation [34]. These data suggest that a reduction of agonist efficacy results in significantly less dromotropic effects of compounds. Therefore, partial A1 agonists seem to be not suited as a therapeutic option for atrial fibrillation. But the reduced dromotropic activity of a partial A1 receptor agonist displays a great safety advantage in patients in which further reduction of heart rate on top of standard therapies is not desirable. Partial A1 agonists have the opportunity of slowing AV conduction only to a certain degree, thereby reducing heart rate without the risk of generating a third degree AV block [35]. An AV block by a full A1 receptor agonist bears a high risk. When heart rate depends only on the ventricular frequency (about 40 beats/min), a life-threatening condition in at-risk patients with already reduced heart function occurs.
Less dependency on the partial character of an A1 receptor agonist is seen for cardioprotective and lipid modulating effects. CVT-3619 [24] inhibits lipolysis and lowers plasma free fatty acids without any cardiovascular effects [25]. VCP28 shows cardioprotection at concentrations that do not reduce heart rate [26]. Capadenoson reduces IS by a preconditioning mechanism at low doses but never evokes an AV block even at maximal dosages.
Cardioprotection from ischemia-induced tissue injury by preconditioning is the most powerful endogenous mechanism for protecting the heart during prolonged ischemic episodes [36]. This might be a new therapeutic and prophylactic option for patients with or at high risk for an acute coronary syndrome.
Preconditioning can be induced by short episodes of recurrent sub-lethal ischemia followed by reperfusion phases, which protects the myocardium from tissue damage during more prolonged ischemic episodes. The cardioprotective effect of preconditioning has been shown in several animal models of acute myocardial ischemia. Endogenous and exogenous stimulation of adenosine A1 receptors has been shown in several animal studies to be involved in this protective effect and in the reduction in IS [37–39]. Activation of A1 receptors results in the subsequent modulation of several downstream signaling cascades [40, 41]. Furthermore, repeated application of A1 agonists leads to a chronic preconditioned state [42].
Peart and Gross described that the full A1 agonist CCPA reduced IS by 36% in a model of ischemia/reperfusion injury in rats [43]. This degree of IS reduction is similar as described here for the partial A1 agonist capadenoson, underlining that the partial character of the compound does not result in reduced cardioprotection.
Besides acute coronary syndrome, coronary heart disease itself might be an indication with beneficial effects of partial A1 agonists. Coronary heart disease is the most significant cause of death in industrial countries. Angina pectoris is a discomfort in the chest caused by myocardial ischemia brought on by exertion. Myocardial ischemia is caused by a transient imbalance between myocardial oxygen demand and supply resulting from a stenosis due to an atherosclerotic plaque. Activation of A1 receptors by a partial A1 agonist can result in a reduction in oxygen consumption by cardioprotective mechanisms and a slightly reduced increase of heart rate at exercise when most angina attacks occur. Partial adenosine A1 receptor agonists can therefore offer a new therapeutic opportunity for the treatment of angina pectoris. The efficacy of a partial A1 agonist in angina pectoris treatment has been proven in a first clinical trial with capadenoson [44]. Furthermore, it was shown that the non-adenosine-like A1 agonist capadenoson have a favorable PK profile with a half-life of 20 h in men [44].
The mechanisms involved in the different modes of actions of partial A1 agonists are not completely understood. Different endogenous adenosine tissue levels and different levels of receptor expression in the heart and other organs might be the key for the effects. High expression levels and a substantial receptor reserve can lead to a low number of receptors needed to be activated by an agonist to evoke a signal whereas tissues with low expression levels and little receptor reserve might need a high number of activated receptors to respond to receptor activation.
These differences are already known for adenosine A1 receptors. In guinea pig atrial myocyctes, it has been shown that there is little receptor reserve for direct activation of potassium current by adenosine, as half-maximal and near-maximal responses are associated with occupancies of 40% and 98% of total A1 receptors. In contrast, there is substantial receptor reserve for indirect inhibition by adenosine of isoproterenol-stimulated calcium current, as half-maximal and near-maximal responses are associated with occupancies of only 4% and 70% of A1 receptors, respectively [23]. Similar effects for G protein-coupled receptors are also known for adrenergic and muscarinic receptor signaling [2]. Furthermore, adipocytes have a very high receptor reserve, which might be an explanation why even partial A1 agonists still show lipolytic effects without any cardiovascular activity [45]. Based on these data, a partial A1 agonist could activate signaling pathways in tissues with a high receptor reserve, but not in tissues with a low one and thereby lead to tissue selective pharmacological effects.
In summary, the importance of developing a drug for cardioprotection by pharmacologically induced preconditioning is well accepted. A1 receptor agonists are promising candidates to fulfill this need, but their broad pharmacological effects on different organs make it difficult to use them as a therapeutic option. Partial A1 agonists have the ability to be tailored to the desired pharmacological effects on cardiovascular diseases with diminished effects on, e.g., bradycardia. Further research is needed to fully evaluate the therapeutic potential of partial adenosine A1 agonists and to evaluate the mechanism of action involved in the differentiated tissue specific signaling pathways of A1 receptors.
Abbreviations
- ACS:
-
Acute coronary syndrome
- CCPA:
-
2-Chloro-N6-cyclopentyladenosine
- CNS:
-
Central nervous system
References
Linden J (2005) Adenosine in tissue protection and tissue regeneration. Mol Pharmacol 67:1385–1387. doi:10.1124/mol.105.011783
Brodde O-E, Michel MC (1999) Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 51:651–690
Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 264:527–552
Mustafa SJ, Morrison RR, Teng B, Pelleg A (2009) Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol 193:161–188. doi:10.1007/978-3-540-89615-9_6
Hussain T, Mustafa SJ (1995) Binding of A1 adenosine receptor ligand [3H]8-cyclopentyl-1,3-dipropylxanthine in coronary smooth muscle. Circ Res 77:194–198
Musser B, Morgan ME, Leid M, Murray TF, Linden J, Vestal RE (1993) Species comparison of adenosine and beta-adrenoceptors in mammalian atrial and ventricular myocardium. Eur J Pharmacol 246:105–111
Van Calker D, Müller M, Hamprecht B (1978) Adenosine inhibits the accumulation of cyclic AMP in cultured brain cells. Nature 276:839–841
Londos C, Cooper DMF, Wolff J (1980) Subclasses of external adenosine receptors. Proc Natl Acad Sci U S A 77:2551–2554
Finegan BA, Lopaschuk GD, Gandhi M, Clanachan AS (1996) Inhibition of glycolysis and enhanced mechanical function of working rat hearts as a result of adenosine A1 receptor stimulation during reperfusion following ischaemia. Br J Pharmacol 118:355–363
Peart J, Headrick JP (2000) Intrinsic A(1) adenosine receptor activation during ischemia or reperfusion improves recovery in mouse hearts. Am J Physiol Heart Circ Physiol 279:H2166–H2175
Headrick JP, Hack B, Ashton KJ (2003) Acute adenosinergic cardioprotection in ischemic-reperfused hearts. Am J Physiol Heart Circ Physiol 285:H1797–H1819. doi:10.1152/ajpheart.00407.2003285/5/H1797
Reichelt ME, Willems L, Molina JG, Sun CX, Noble JC, Ashton KJ, Schnermann J, Blackburn MR, Headrick JP (2005) Genetic deletion of the A1 adenosine receptor limits myocardial ischemic tolerance. Circ Res 96:363–367. doi:10.1161/01.RES.0000156075.00127.C3
Ely SW, Berne RM (1992) Protective effects of adenosine in myocardial ischemia. Circulation 85:893–904
Mubagwa K, Flameng W (2001) Adenosine, adenosine receptors and myocardial protection: an updated overview. Cardiovasc Res 52:25–39
Liao Y, Takashima S, Asano Y, Asakura M, Ogai A, Shintani Y, Minamino T, Asanuma H, Sanada S, Kim J, Ogita H, Tomoike H, Hori M, Kitakaze M (2003) Activation of adenosine A1 receptor attenuates cardiac hypertrophy and prevents heart failure in murine left ventricular pressure-overload model. Circ Res 93:759–766. doi:10.1161/01.RES.0000094744.88220.62
Stone TW, Ceruti S, Abbracchio MP (2009) Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol 193:535–587. doi:10.1007/978-3-540-89615-9_17
Liang BT, Donovan LA (1990) Differential desensitization of A1 adenosine receptor-mediated inhibition of cardiac myocyte contractility and adenylate cyclase activity. Relation to the regulation of receptor affinity and density. Circ Res 67:406–414
Green A (1987) Adenosine receptor down-regulation and insulin resistance following prolonged incubation of adipocytes with an A1 adenosine receptor agonist. J Biol Chem 262:15702–15707
Lee HT, Thompson CI, Hernandez A, Lewy JL, Belloni FL (1993) Cardiac desensitization to adenosine analogues after prolonged R-PIA infusion in vivo. Am J Physiol 265:H1916–H1927
Roman V, Keijser JN, Luiten PG, Meerlo P (2008) Repetitive stimulation of adenosine A1 receptors in vivo: changes in receptor numbers, G-proteins and A1 receptor agonist-induced hypothermia. Brain Res 29:69–74. doi:10.1016/j.brainres.2007.11.044
Srinivas M, Shryock JC, Dennis DM, Baker SP, Belardinelli L (1997) Differential A1 adenosine receptor reserve for two actions of adenosine on guinea pig atrial myocytes. Mol Pharmacol 52:683–691
Parsons WJ, Stiles GL (1987) Heterologous desensitization of the inhibitory A1 adenosine receptor-adenylate cyclase system in rat adipocytes. Regulation of both Ns and Ni. J Biol Chem 262:841–847
Longabaugh JP, Didsbury J, Spiegel A, Stiles GL (1989) Modification of the rat adipocyte A1 adenosine receptor-adenylate cyclase system during chronic exposure to an A1 adenosine receptor agonist: alterations in the quantity of GS alpha and Gi alpha are not associated with changes in their mRNAs. Mol Pharmacol 36:681–688
Fatholahi M, Xiang Y, Wu Y, Li Y, Wu L, Dhalla AK, Belardinelli L, Shryock JC (2006) A novel partial agonist of the A1-adenosine receptor and evidence of receptor homogeneity in adipocytes. J Pharmacol Exp Ther 317:676–684. doi:10.1124/jpet.105.099119
Dhalla A, Santikul M, Smith M, Wong MY, Shryock J, Belardinelli L (2007) Anti-lipolytic activity of a novel partial A1 adenosine receptor agonist devoid of cardiovascular effects: comparison with nicotinic acid. J Pharmacol Exp Ther 321:327–333. doi:10.1124/jpet.106.114421
Urmaliya VB, Pouton CW, Devine SM, Haynes JM, Warfe L, Scammelis PJ, White PJ (2010) A novel highly selective adenosine A1 receptor agonist VCP28 reduces ischemia injury in a cardiac cell line and ischemia–reperfusion injury in isolated rat hearts at concentrations that do not affect heart rate. J Cardiovasc Pharmacol 56:282–292. doi:10.1097/FJC.0b013e3181eb8563
Kiesmann WF, Elzain E, Zablocki F (2009) A1 adenosine receptor antagonists, agonists and allosteric enhancers. In: Wilson CN, Mustafa SJ (eds) Handbook of experimental pharmacology 193. Springer, Berlin, pp 25–58
Nell P, Albrecht-Küpper B (2009) The adenosine A1 receptor and its ligands. In: Lawton G, Witty DR (eds) Progress in medicinal chemistry. Elsevier, Amsterdam, pp 164–201. doi:10.1016/S0079-6468(08)00204-X
Lorenzen A, Fuss M, Vogt H, Schwabe U (1993) Measurement of guanine nucleotide-binding protein activation by A1 adenosine receptor agonists in bovine brain membranes: stimulation of guanosine-5′-O-(3[35S]thio)triphosphate binding. Mol Pharmacol 44:115–123
Lorenzen A, Guerra L, Vogt H, Schwabe U (1996) Interaction of full and partial agonists of the a1 adenosine receptor with receptor/G protein complexes in rat brain membranes. Mol Pharmacol 49:915–926
Rosentreter U, Krämer T, Shimada M, Hübsch W, Diedrichs N, Krahn T, Henninger K, Stasch JP, Wischnat R (2003) PCT Int Appl, WO 03 053441 CAN 139:36542
Wu L, Belardinelli L, Zablocki JA, Palle V, Shryock JC (2001) A partial agonist of the A(1)-adenosine receptor selectively slows AV conduction in guinea pig hearts. Am J Physiol 280:H334–H343
Aderis Pharmaceuticals. Safety and efficacy study of an A1-adenosine receptor agonist to slow heart rate in atrial fibrillation 2005. Available from: http//:www.clinicalstrials.gov./ct/show/NCT00040001?order=1
Bayer Schering Pharma AG (2007) Clinical study in ClinicalTrials.gov identifier NCT00568945. Bayer Schering Pharma AG, Wuppertal
Zablocki JA, Wu L, Shryock JC, Belardinelli L (2004) Partial A1 adenosine receptor agonists from a molecular perspective and their potential use as chronic ventricular rate control agents during atrial fibrillation (AF). Curr Top Med Chem 4:839–854
Otani H (2008) Ischemic preconditioning: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 10:207–247. doi:10.1089/ars.2007.1679
Baxter GF (2002) Role of adenosine in delayed preconditioning of myocardium. Cardiovasc Res 55:483–494
Thornton JD, Liu GS, Olsson RA, Downey JM (1992) Intravenous pretreatment with A1-selective adenosine analogues protects the heart against infarction. Circulation 85:659–665
Headrick JP, Lasley RD (2009) Adenosine receptors and reperfusion injury of the heart. Handb Exp Pharmacol 193:189–214. doi:10.1007/978-3-540-89615-9_7
Sommerschild HT, Kirkebøen KA (2002) Preconditioning—endogenous defence mechanisms of the heart. Acta Anaesthesiol Scand 46:123–137
Hausenloy DJ, Yellon DM (2006) Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 70:240–253. doi:10.1016/j.cardiores.2006.01.017
Dana A, Baxter GF, Walker JM, Yellon DM (1998) Prolonging the delayed phase of myocardial protection: repetitive adenosine A1 receptor activation maintains rabbit myocardium in a preconditioned state. J Am Coll Cardiol 31:1142–1149
Peart JN, Gross GJ (2003) Adenosine and opioid receptor-mediated cardioprotection in the rat: evidence for cross-talk between receptors. Am J Physiol Heart Circ Physiol 285:H81–H89. doi:10.1152/ajpheart.00985.200200985.2002
Tendera M, Sosnowski M, Gaszewska-Zurek E, Parma Z, Neuser D, Seman L (2007) The oral adenosine A1 receptor agonist, BAY 68-4986, reduces heart rate at peak exercise in patients with chronic stable angina. J Am Coll Cardiol 49(suppl A):232A
Dhalla AK, Shryock JC, Shreeniwas R, Belardinelli L (2003) Pharmacology and therapeutic applications of A1 adenosine receptor ligands. Curr Top Med Chem 3:369–385
Acknowledgments
The authors would like to thank Raimund Kast and Katja Zimmermann for the data on the 35S-GTP binding assay and the Langendorff heart testing of capadenoson.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Albrecht-Küpper, B.E., Leineweber, K. & Nell, P.G. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signalling 8 (Suppl 1), 91–99 (2012). https://doi.org/10.1007/s11302-011-9274-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-011-9274-3