
Abstract
Palm oil has a balanced fatty acid composition and has no trans fat. As a result, its use in food has increased as food-labeling laws have changed to specify trans fat content. Increasing oil production is the main goal in oil palm breeding. Genetic mapping and genomic studies in palm trees are necessary to understand the genetic architecture of economic traits of importance for palm oil production. To help achieve this, we sampled 422 oil palms from MPOB (Malaysian Palm Oil Board)Angola germplasm collection and measured 13 economic traits from these palms. Multi-locus genome-wide association studies (GWAS) were conducted using least absolute shrinkage and selection operator (LASSO) and genome-wide efficient mixed model analysis. We identified 19 quantitative trait loci (QTLs) for 8 traits. Of these, four Angola-specific QTLs associated with bunch components were detected on chromosomes 4, 8, and 11. These QTLs are potentially useful for introgression of desirable genes from the Angola palms to advanced breeding populations for improvement of bunch and oil yield traits. The majority of the QTLs were detected by LASSO-A, in which the p values of individual markers were calculated based on bootstrapped standard errors. Many of the detected QTLs are nearby known QTLs detected from linkage studies reported by other research groups. We also conducted genomic selection (GS) for the 13 traits and concluded that GS can be an effective tool for oil palm breeding. This is the first GWAS and GS study conducted on oil palm germplasm from Angola, and the results can be very useful in oil palm genetic studies and breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
One of the prime objectives in oil palm breeding is to improve oil yield. Over the past six decades, the average palm mesocarp oil yield has increased from ~ 1.0 to 3.0 t per hectare. In Southeast Asia, early improvement efforts largely used descendants of four dura palms established in the Bogor Botanical Garden, Java. Oil palm breeders then made use of limited numbers of pisiferas from Ekona, Calabar, Yangambi, La Me, and AVROS to generate a commercial hybrid, tenera. However, the few palms originally used for breeding offered very little genetic diversity. Thus, Central and Western Africa (home of the oil palm) were prospected for more germplasms (Rajanaidu and Jalani 1994). These materials have great potential for oil palm improvement as they have a large genetic diversity and possess new traits, such as less saturated oil, lower height increment, long stalk, high kernel, carotene, oleic acid, and vitamin E contents (Kushairi et al. 2011).
Malaysian Palm Oil Board (MPOB, previously called MARDI and PORIM) initiated several projects to search and collect oil palm germplasms from the primary center of diversity of natural oil palm that is in Africa. One of the countries explored for the germplasm collection is Angola. In this country, about 54 bunches were collected from eight sites (Rajanaidu et al. 1991) in 1991 and were planted at the MPOB research station in Kluang, Johore to assess their phenotypic performance. Field evaluation of these palms demonstrated their potential for improving stalk length, oil yield, height increment, and oil unsaturation (Kushairi et al. 2003; Noh et al. 2002). Some palms have excellent fruit quality traits, comparable to the advanced female parental lines (duras) currently used to produce commercial planting materials, e.g., large fruit size and good fruit-to-bunch (FTB) ratio, which, in combination, can give high mesocarp oil yield. These palms are comparable to advanced female parental lines (duras) currently used to produce commercial planting materials. In terms of height increment, materials from sites 7 and 8, located in the north, were reportedly shorter (height increment of 20 to 30 cm/year) than those from the south of Angola, sites 1, 2, and 3 (50 to 60 cm/year).
One of the major developments in plant and animal genetic research is the application of molecular markers for detection of DNA polymorphism, which could be associated with traits of interest. The use of molecular markers has enabled the study of mutations and recombination events in a species or population to be uncovered. Significant efforts at exploiting recombination events for linkage studies and, subsequently, the discovery of markers associated with traits of interest have been reported. In mapping populations, only a small fraction of quantitative trait loci (QTLs) associated with the traits have been identified. Furthermore, due to the relatively low recombination events among hybrid crosses than in wild or unadapted populations, any QTL detected can be loosely associated and are often population specific. Nevertheless, linkage mapping does offer the power to identify QTLs with large genetic effects.
The linkage mapping approach has allowed detection of major QTLs associated with important traits in oil palm, such as height and fatty acid composition (FAC) (Lee et al. 2015; Montoya et al. 2013; Pootakham et al. 2015; Singh et al. 2009; Ting et al. 2014). The QTLs for these traits explained 10 to 51% of the phenotypic variation. In 2010, using several mapping populations, Billotte et al. (2010) reported QTLs associated with yield parameters (bunch number, bunch weight, fresh fruit bunch yield) and bunch components (fruit weight, ratios of FTB, e.g., mesocarp-to-fruit and kernel-to-fruit) in oil palm. As not all the QTLs influencing a particular trait are present in any specific population, validating these QTLs poses a great challenge. The introduction of an alternative method to detect QTLs for phenotypic traits in plant species has opened up a promising opportunity for validation as well as identification of additional QTLs. The method, known as association mapping (AM) analysis, was initially used in human genetic research due to the limited number of progenies per family available for analysis, which has a similar limitation faced in large plant species, like oil palm. The AM method involves phenotypic and genotypic characterizations of unadapted or wild populations, followed by statistical association analysis between the genotype polymorphism and the phenotypic trait variation. In such populations, relatively more QTLs underlying a trait are present. These populations carry numerous recombination events, which optimize the detection of QTLs with minor effects and efficiently eliminate loosely associated QTLs. This feature ideally complements the genetic linkage mapping approach. The AM analysis was carried out successfully for oil palm recently, which led to the identification of loci linked to mesocarp oil content (Teh et al. 2016) and shell-to-fruit (STF) ratio (Kwong et al. 2016).
In AM, the samples investigated often include individuals from diverse genetic backgrounds, different selection histories, and diverse geographic origins. These samples often exhibit some degrees of structure or subdivision, which can potentially cause false positive or false negative associations (Pritchard et al. 2000). Even oil palm germplasm samples collected from one country or region exhibit population structure (Ong et al. 2015). The covariates arising from the presence of subgrouping among the samples bring about bias in estimating allelic effects in AM and, subsequently, generate false-positive associations (Kennedy et al. 1992). Analysis methods that include population structure in AM have been introduced (Yu and Buckler 2006) to help minimize false positives. Principal component analysis (PCA) and the corresponding Q matrix generated have been used to capture population structure effects in linear mixed model association analysis. The false positive rate can be further reduced by incorporating the relationships among individual samples, i.e., incorporating the kinship (K) matrix into the linear mixed model (Yu and Buckler 2006).
Genome-wide association studies (GWAS) are now considered as a routine procedure for QTL detection in wild populations and diverse panels of agricultural species. The Q+K mixed model introduced above is a way to alleviate heavy computational burden when the number of markers is huge and is not necessarily the best method. The optimal method should be one of the multiple marker approaches (Li et al. 2011; Segura et al. 2012; Tian et al. 2011; Waldmann et al. 2013; Zhang et al. 2011) that fit multiple markers in a single model. However, these multiple marker models cannot handle unlimited number of markers and we are forced to use the Q+K model if the number of markers is huge, say > 100 k, because it provides a convenient way to scan the genome by fitting one marker in the model at a time. In reality, many GWAS populations may not have extremely highdensity marker maps. After excluding lowquality markers and markers with rare frequencies, the number of available markers may only be in the order of a few thousand.
The main objectives of the present work are, first, to study the population structure of the Angola populations by means of the SNP markers, and secondly, to carry out association analysis using least absolute shrinkage and selection operator (LASSO) and genome-wide efficient mixed model analysis (GEMMA) methods. This is our first attempt in trying to detect QTLs for economically important traits in oil palm using germplasm populations as compared to previous reports on oil palm utilizing advanced or introgressed populations.
Materials and methods
Plant materials and phenotype collection
The MPOB-Angola germplasm was evaluated for 52 phenotypic traits, which are divided into three categories namely, yield recording, bunch analysis, and vegetative parameters. Yield recording and bunch analysis (Blaak et al. 1963) were initiated in 1997 and took up to 8 years to complete. The vegetative measurements were conducted in 2001, seventh year after planting based on standard procedure (Corley et al. 1971). The phenotypic data were applied in the principal component analysis (PCA). The results (data not shown) were used to select the Angola populations used in this study as they showed wide distribution in the PCA analysis. Young unopened leaves were harvested from 422 palms originating from six sites in Angola. The number of palms sampled per site is summarized in Supplementary Table S1. The number was uneven due to plant mortality during germination and culture in the nursery (Kushairi et al. 2003).
In this study, three vegetative measurements (rachis length, height, and height increment) and ten bunch analysis traits were applied to association studies. The abbreviations and full names of the traits analyzed are given in Supplementary Table S2. Pearson correlations among all the traits were estimated using the SPSS 16.0 statistical software.
DNA extraction and genotyping
Genomic DNA was extracted from young leaves using the modified CTAB method (Doyle 1990). DNA concentration and purity were determined using the NanoDrop spectrophotometer (NanoDrop Technologies Inc.). Prior to the SNP assay using the Illumina platform, the DNA was re-quantified using the Quant-iT™ PicoGreen® dsDNA Reagent (Invitrogen) and subsequently normalized to 50 ng/μl.
The oil palm OPSNP3 array (Ting et al. 2014) that contained 4451 markers was used to genotype 422 DNA samples on the Illumina Infinium II Bead Chip platform. The sequence information of the SNPs is available at the Genomesawit website (http://genomsawit.mpob.gov.my). SNP hybridization assay was carried out following the Illumina’s protocol. The SNPs were discovered using fluorescent staining (green and red) and detected by the Illumina Bead Array Scanner.
SNP data preparation
The SNP genotyping data were extracted using the GenomeStudio® Data Analysis Software (Illumina Inc.). For our analysis, only the genotype data of the mapped SNPs were extracted (Ting et al. 2014). Prior to any analysis, markers that had > 10% missing data were excluded. The data were organized to allow analysis using PowerMarker (Liu and Muse 2005) and Structure (Pritchard et al. 2000). PowerMarker was used to compute the allelic frequencies, which were then used to discard monomorphic markers and markers with minor allele frequency < 0.05. Prior to GWAS, we tested the population structure of the data using the Structure software (Pritchard et al. 2000). The K value (number of components) varied from 2 to 10, and each K value was tested using five independent runs. For each K value, the first 10,000 Markov chain Monte Carlo (MCMC) iterations were deleted (burn-in deletion), and an additional 10,000 iterations were then collected as observations in the posterior sample. Once the K value was determined, the same software was used to calculate the population membership for each individual. This information was stored in a Q-matrix and included as fixed effects in the Q+K model of GWAS. Principal component analysis and k-mean cluster analysis were also carried out to generate design matrices to control the population structure effects. The results using different structures were compared.
Multiple locus models for genome-wide association studies
The LASSO method was used and implemented in an R package called GLMNET/R (Friedman et al. 2010). Unfortunately, the package does not have a mechanism to calculate the p value of each marker. A newly released R package called covTest (Lockhart et al. 2014) was particularly designed for p value calculation. However, it only applies to situations where the number of markers is less than the sample size, which is not the case of this experiment. We proposed two methods to calculate the p values for LASSOestimated marker effects. One is the bootstrap method (Efron and Tibshirani 1994), and the other is the method of best linear unbiased prediction (BLUP) equation (Henderson 1975). With these methods, we estimated the variance of each estimated marker effect and then calculated a Wald test statistic by dividing the squared estimated effect by the variance of the estimated effect. This test is approximated by a chi-square distribution under the null model so that the p value can be computed from this chi-square distribution.
For a data set with m = 1081 markers and n = 422 individuals, a multiple locus model may work better than the genome scanning approaches. The LASSO method (Tibshirani 1996) is the most commonly used method for variable selection, and we investigated the applicability of LASSO to GWAS using this data set. The LASSO estimates of marker effects (m × 1 vector) are defined as follows:
where y is an n × 1 vector of the phenotypic values of a trait measured from all palm trees. Other terms are defined as follows: β 0 is the intercept (grand mean); X k is an n × 1 vector of genotype indicator variables for marker k where an element of this vector is defined as 1, 0, or − 1, respectively, for the three genotypes, say AA, AG, or GG; β k is the effect of marker k; λ is a shrinkage parameter determined via 10-fold cross-validation to minimize the mean squared errors (MSEs). The first term in Eq. (1) defines the model goodness of fit and the second term is an L 1 penalty. The GLMNET/R program (Friedman et al. 2010) was used to perform the LASSO estimation of parameters. Unfortunately, the software package does not provide a mechanism to calculate the estimation error for each marker effect and thus no p value is attached to a marker. The covTest package (Lockhart et al. 2014) is able to calculate the p values for the LASSO estimates, but it only applies to the situation where n > m, which is certainly not the case in this study. We adopted two approaches to calculate the estimation error for \( {\widehat{\beta}}_k \). The first approach is the bootstrap method (Efron and Tibshirani 1994) where 1000 bootstrap samples were drawn and analyzed to obtain an empirical variance whose square root is the standard error of an estimated marker effect. We calculate the Wald test statistic defined as
The p value for marker k is obtained from the Wald test
which follows approximately a chi-square distribution with one degree of freedom under the null hypothesis. The second approach is an approximation via Henderson’s mixed model equation (Henderson 1975). Let S be the number of markers with non-zero effects after LASSO variable selection, where S < m. Define the LASSO estimated effects for these selected markers by \( {\widehat{\beta}}_j \) for j = 1 , … , S. The Henderson mixed model equation for these selected marker effects can be approximated by
where X 0 is an n × 1 vector of unity. These mixed model equations assume that \( \widehat{\beta} \) are the posterior means (BLUP) of marker effects with a prior variance of each regression coefficient defined as \( {\sigma}_{\beta_j}^2 \). This prior variance is unknown but may be replaced by \( {\widehat{\beta}}_j^2 \). In other words, we let \( {\sigma}_{\beta_j}^2={\widehat{\beta}}_j^2 \) and obtain the following approximate variance-covariance matrix of \( \widehat{\beta} \)
where \( {\widehat{\sigma}}^2 \) is the estimated residual variance. In compact matrix notation, we can define \( \operatorname{var}\left(\widehat{\beta}\right)=C{\widehat{\sigma}}^2 \) where C is the square matrix of Eq. (5) that excludes \( {\widehat{\sigma}}^2 \). Define the jth row and the jth column of matrix C by C jj , we have \( \operatorname{var}\left({\widehat{\beta}}_j\right)={C}_{jj}{\widehat{\sigma}}^2 \), which facilitates a Wald test shown in Eq. (2). This approximation may not be as good as the bootstrap method, but it is computationally much more efficient.
For the multiple locus models, there is no need for Bonferroni correction because the estimated effects are already shrunken towards zero. Therefore, the critical value of 0.05 can be used to declare statistical significances for all markers.
Simulation experiments
To investigate the powers and type 1 errors of the proposed new multiple locus methods, we carried out replicated simulation experiments using the linkage map constructed by Ting et al. (2014) and genotypes of the 422 oil palms and assigned 20 loci as QTLs. The heritability contributed by the 20 simulated QTLs was set at 50%. A random error was sampled from a normal distribution with mean zero and variance σ 2, which was calculated according to the heritability and the genetic variances. Locations and effects of the 20 QTLs are shown in Table 1. The QTL effect ranges from 0.5 to 8.0% of the phenotypic variance. The simulation was replicated 200 times. The power of QTL is defined as the proportion of replicates where the p value of the QTL is less than 0.05 for the multiple locus models. For the classical GWAS models, the p value criterion of positive detection is set as p being less than 0.05/m = 0.05/1081 = 0.00004625 (after Bonferroni correction for multiple tests). The type 1 error of a method is defined as the proportion of false positives out of all markers not assigned a QTL effect. In summary, we examined two new methods, LASSO with bootstrapped error calculation (LASSO-A) and LASSO with BLUP error calculation (LASSO-B), and one existing method, the GEMMA (Zhou and Stephens 2012). The three aforementioned methods were also used for the real data analysis.
Mapping of markers on EG5 genomes build
A total of 54 markers (Supplementary Table S3) associated with selected bunch components and vegetative traits detected here as well as other publications (Billotte et al. 2010; Jeennor and Volkaert 2014; Kwong et al. 2016; Teh et al. 2016) were mapped onto the EG5 genome build (Singh et al. 2013) published in the GenomSawit website. (http://genomsawit.mpob.gov.my/genomsawit/). Briefly, the PCR primer sequences of simple sequence repeat (SSR) markers were retrieved from the respective publications and CIRAD website (http://tropgenedb.cirad.fr/tropgene/JSP/interface.jsp?module=OILPALM). Electronic PCR (e-PCR) of these primers was carried on oil palm genome EG5 build by allowing four mismatches and two gaps of the primers, with 50–3000 bp amplicon size, using software e-PCR v2.3.11. The accession number for some of these SSR markers was retrieved from Corsat web (www.corsat.agr.ku.ac.th/doc/oilpalm/256.xls), and their sequences were retrieved from NCBI database. MPOB SNP marker sequences (Ting et al. 2014) were retrieved from the GenomSawit website. Due to differences between genome sequence deposited in public database and that in GenomSawit web, SNP markers that were reported based on genome sequence (CM002082.1, CM002085.1, and CM002090.1) including the 120 bp upstream and downstream sequences of the SNPs were retrieved from the respective chromosomes. All sequences were BLASTn (v2.2.26+) search against EG5 build. Coordinates of ePCR product and BLAST search were used to identify position of these markers on EG5 build. Circos plot (Krzywinski et al. 2009) was used to visualize the location of the markers.
Results
SNP data cleaning
Of the 4451 SNPs genotyped across the 422 samples, we shortlisted 1210 SNPs as these SNPs were successfully mapped into genetic linkage groups described by Ting et al. (2014). We have applied the marker positions established in these linkage groups for AM analysis. Of the 1210 SNPs, 48 were monomorphic and 24 had > 10% missing data across our samples. Further, the allelic frequencies revealed another 57 SNPs being rare alleles (< 0.05 frequency). As a result, a total of 129 markers were removed while the remaining 1081 SNPs were eventually used for structure determination and association analysis.
Power and type 1 error analysis
The empirical powers and type 1 errors obtained from 200 replicated simulations for the 20 QTLs are listed in Table 1. LASSO-A had perfect power for almost all QTL, even for QTL with very small effects (explaining 0.5% of the phenotypic variance), while LASSO-B had lower power compared to LASSO-A. GEMMA had the lowest power among the three methods. The conclusion is that the multiple locus methods (LASSO-A and LASSO-B) are more powerful than the single marker scanning method (GEMMA). Surprisingly, the type 1 error for LASSO-A was zero (0.000) for all loci and LASSO-B had the highest type 1 error (0.002). In general, all methods have type 1 errors under control.
Population structure
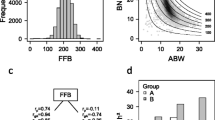
We also used the 1081 markers to calculate a genetic distance matrix between all pairs of oil palms and then draw an unrooted phylogeny as shown in Fig. 1a. The 422 oil palms appear to have 17 major clusters. One of the clusters can be further divided into several subgroups. This number of clusters matches closely the number of families, which happens to be 19. We also performed k-mean cluster analysis and principal component analysis. The second principal component is plotted against the first principal components, as shown in Fig. 1b, where three clusters appear to be evident but they do not match the three clusters (color coded) obtained from the k-mean cluster analysis. Figure 1c shows the plot of the percentage contribution of individual component against the number of principal components. The first component only contributes about 2.75% of the total variation of the markers. The second component contributes 2.65%. The top 18 components cumulatively contribute about 30% of the total marker variation. This means that the population structure cannot be explained by a few principal components. Figure 1d shows the plot of within cluster mean square error against the number of components drawn from the k-mean cluster analysis. Again, it takes about 15–20 clusters to significantly reduce the MSE, implying that there are too many clusters to capture the population structure. We also used the BIC proposed by Fraley and Raftery (2007) to determine the number of clusters. The result indicated that the cluster number should be 15. Results of the k-mean cluster analysis and phylogenetic analysis are very consistent in a sense that the majority of the clusters from the k-mean analysis are uniquely placed in specific clades of the phylogenetic tree.

Population structure analysis. a Unrooted phylogeny of the 422 accessions of oil palms. b Principal component plot, where the three clusters obtained from the k-mean cluster analysis are color coded. c Percentage contributions of principal components plotted against the number of principal components. d Within cluster mean squared error from the k-mean cluster analysis plotted against the number of clusters
Correlation analysis among traits
Table 2 shows the pairwise Pearson correlations that have reached the 0.05 significant level. Generally, negative correlations were observed between the kernel factors (kernel-to-bunch (KTB), kernel-to-fruit (KTF), and kernel yield (KY)) and oil-related components (mean fruit weight (MFW), mean nut weight (MNW), mesocarp-to-fruit (MTF), oilto-bunch (OTB), and oil yield (OY)). The correlation among the kernel factor traits was positive. Similarly, positive correlation was attained between oilrelated trait components. The vegetative traits (rachis length (RL), height increment (HI), height (HT)) showed significant positive correlations with most of the oil-related traits (e.g., MFW, MTF, OTB, OY) but were negative with KTF.
Association of SNPs with selected traits
Manhattan plots of all the traits for the three GWAS methods are shown in Fig. 2 and Supplementary Figs. S1 and S2. We identified 19 QTLs for eight agronomic traits of oil palm (HI, HT, KTB, MTF, MFW, OTB, OY, and RL) from the three GWAS methods. None of the methods detect any QTL associated with the five remaining traits (FTB, KTF, KY, MNW, and STF). LASSO-B detected the most QTLs (13), followed by LASSO-A (10) and GEMMA, which only detected three. Three QTLs were detected by all models while another three were identified by LASSO-A and LASSO-B. One QTL appeared to control more than one trait (pleiotropic effect), e.g., SNPM02805, which had a significant effect on three related traits (OTB, MFW, and MTF). Of the 13 traits, we only present the results for 12 and KY was omitted as it had an estimated effect of zero for all markers from all the three methods (results not shown). Detailed information on the significantly associated QTLs is summarized in Table 3.

Manhattan plots of the first set of four traits resulting from three GWAS methods. The four traits are labeled as FTB (fruittobunch), HI (height increment), HT (height), and KTB (kerneltobunch). The three GWAS methods are LASSO-A, LASSO-B, and GEMMA. The dashed horizontal line in each Manhattan plot depicts the significance threshold
Of the 19 significant QTLs detected by LASSO, 13 were associated with the bunch quality components and the remaining six to the vegetative traits. The 13 QTLs influencing bunch components were detected on eight chromosomes (3, 4, 5, 6, 7, 8, 11, and 15). Six QTLs associated with vegetative parameters were found on five chromosomes (2, 4, 7, 10, and 16). QTLs influencing both bunch components and vegetative traits were detected on linkage groups (LGs) 4 and 7. We carried out further analysis to determine the effect of marker genotypes on the traits concerned. The results are summarized in Supplementary Table S4. Significant effects (p < 0.05) were attained for the different genotypes of all QTLs on their respective traits. Different genotype classes of a QTL (SNPM02805) also showed significant effect for three traits—MFW, MTF, and OTB. Boxplot was prepared using R package to visualize the effects of the three genotypes on the means of these traits (Fig. 3).

Effects of genotypes GG, GC, and CC of SNPM02805 on MFW (mean fruit weight), MTF (mesocarptofruit), and OTB (oiltobunch). The means of the traits for each genotype class were significantly different at p < 0.05
Surprisingly, the largest QTL detected by LASSO-A for trait RL only contributed 1.48% of the phenotypic variance. The smallest QTL detected by LASSO-A for trait MFW explained 0.063% of the phenotypic variance. QTLs with such small effects are not detectable with the classical genome scanning approach. For example, GEMMA only detected three QTL from all the 13 traits. We realized that the R 2 values (proportions of phenotypic variance contributed by QTL) for the QTL detected by LASSO-A and LASSO-B may be severely biased downwardly due to shrinkage. The three largest QTL detected by the LASSO methods were all detected by GEMMA, which showed a much higher proportion contributed by each of the three QTL (> 4%).
Comparison of QTLs
The QTLs identified in this study were also compared to those identified by other research groups. For ease of discussion and comparison, the LGs where QTLs were detected either in this study or other relevant publications were re-assigned based on the EG5 genome build (Singh et al. 2013) and are illustrated in Fig. 4.

Mapping of 54 QTLs associated with selected bunch components and vegetative traits identified in the present study and those reported by four independent groups (Billotte et al. 2010; Jeennor and Volkaert 2014; Teh et al. 2015; Kwong et al. 2016) on EG5 genome build (Singh et al. 2013). The most outer ring are 16 chromosomes of EG5, followed by the markers’ ID and associated traits, locations of the markers on the chromosomes from five different sources, and the linkage group of the markers. The positions of these markers (SNPM0957, SNPM00736, and SNPM02015) could not be determined on EG5 but were roughly estimated based on the EG8 genome build (unpublished) (filled star). The positions of markers SNPM00995 and SNPM01889 could not be located on the EG5 genome build (asterisk). However, these markers were mapped on LGs 1 and 2 according to Ting et al. (2014), which correspond to chromosomes 3 and 8, respectively
Multiple QTLs for bunch components detected here as well as by other research groups were found on chromosomes 3, 5, 6, 7, and 15. On chromosome 3, apart from the QTL linked to MTF detected in this study, Billotte et al. (2010) also reported a QTL for the same traits in nearby regions. Interestingly, the QTL for MTF was also located close to the QTL for a related bunch quality trait, oil-to-fruit (OTF), reported by Jeennor and Volkaert (2014). Chromosome 5 was also interesting as it contained QTLs associated with bunch quality component traits reported by four independent research groups. This study also located a QTL associated with MFW (a bunch quality component trait) in the same chromosome. On chromosome 6, the QTLs for bunch components discovered by three independent research groups were located at two different regions. The QTL linked to MFW detected in this study was in close proximity to related traits, MTF and oil-to-dry-mesocarp (OTDM), described by Billotte et al. (2010). Similarly, the QTLs for bunch components are also found in two regions on chromosome 7. One of the regions contained QTLs for OY and MTF detected in this study, together with a QTL for FTB reported by Jeennor and Volkaert (2014). The QTL in the second region of this chromosome is associated with KTB detected in the current study. The QTLs for bunch components on chromosome 15 were identified in the current analysis as well as that of Billotte et al. (2010). Additionally, new QTLs associated with bunch components were detected in the current study on chromosomes 8 and 11, which might be specific to the Angola germplasm.
Figure 4 reveals three chromosomes (2, 4, and 7) that contain QTLs for both vegetative traits and bunch components. On chromosome 2, the QTLs are separated according to their associated traits, each located at two different regions of the chromosome. The first region contained QTLs for bunch components (STF) reported by Kwong et al. (2016), whereas the second region composed of QTLs linked to the vegetative traits (RL) identified by Billotte et al. (2010). Our study also identified QTL for RL in the nearby region with an additional QTL linked to HI and HT. Billotte et al. (2010) also described some QTLs for RL on chromosome 4, which are close to one of the QTLs associated with MFW, MTF, and OTB detected in this study. We also detected a QTL for RL, but it was positioned at the opposite end of the same chromosome. On chromosome 7, one of the regions contained QTLs for bunch components identified in this study and Jeennor and Volkaert (2014) while the second section composed of QTLs identified in our study for bunch components (KTB) as well as vegetative traits (HT). The QTLs for RL found on chromosome 16 were based on the work carried out here and by Billotte et al. (2010).
Discussion
Our 422 oil palm accessions were genotyped for 4451 SNP makers, but only 1081 were polymorphic. In this case, a multiple marker approach can be easily applied. The most efficient highdimensional multiple regression model is the LASSO method Tibshirani (1996). We decided to adopt this method of association studies for 13 quantitative traits of oil palms and compare the result with the traditional Q+K mixed model analysis implemented in the GEMMA software package (Zhou and Stephens 2012).
This study represents the first GWAS analysis of the Angola oil palm germplasm. Although it is the most important oil producing tree in Malaysia and Indonesia, representing an important commodity worldwide, the crop however has not been extensively studied. Presently, we have obtained 1081 polymorphic markers for the 422 palms. We were able to use a multiple variate GWAS method like LASSO to test all markers simultaneously. The genome scanning approach, such as the GEMMA method, usually requests a multiple test adjustment procedure to ensure overall appropriate type 1 error control. We did not apply the Bonferroni correction because LASSO is a multiple marker model where all markers are fitted to the single model and all effects are estimated and tested simultaneously. As a result, there is no multiple tests issue. In addition, using the 0.05 criterion, our type 1 error (LASSO-A) is still zero. We already had a better control of type 1 error without multiple test correction compared with GEMMA for the LASSO-A method. Not only does LASSO have high power but also have low false positive rate, indicating the better performance of LASSO over GEMMA.
The traditional linear mixed model approach for GWAS (Yu and Buckler 2006) scans the genome one marker at a time, not because it is the best approach but because it can deal with millions of markers in a convenient way. In most GWAS populations, the number of markers may be less than 10 or 20 K. In these situations, LASSO is highly recommended. Unfortunately, the GLMNET program for LASSO estimation does not provide a mechanism to draw the p values for individual markers. We presented two approaches to calculate the p values. The bootstrap approach is very efficient compared to the BLUP approach, but it is computationally more intensive than the latter. Therefore, when the population size is small, the bootstrap approach is recommended while the BLUP method may be applied when the population size is extremely large.
Given the fact that all detected markers have small effects for the 12 traits analyzed, we suspected that all the traits are typical polygenic traits. We therefore performed genomic prediction using all the 1081 markers to see whether or not it is feasible to conduct genomic selection in the oil palm samples analyzed in the present study. We used three methods of genomic selection, LASSO (Tibshirani 1996), BLUP (VanRaden 2008), and BayesB (Meuwissen et al. 2001), and obtained predictabilities for all traits via a 10-fold cross-validation analysis. The predictability is defined as the squared Pearson correlation coefficient between the observed and predicted trait values. The results are listed in Table 4. Among the three methods, BayesB had the highest average predictability (0.1352), followed by the BLUP method (0.1155) and the LASSO method (0.0922). For the BayesB method, the predictability ranged from 0.0377 for KY to 0.2142 for RL. All three vegetative traits had predictabilities higher than 0.20, and the bunch component traits typically had lower predictabilities. Although the predictabilities overall are not very high, they are acceptable and suggest that genomic selection can be an effective tool for oil palm breeding. Note that efficiency of genomic selection depends on two factors, predictability and selection intensity. We can still perform genomic selection when the predictability is not high because we can increase selection intensity by genotyping a large number of seedlings as candidates of selection.
Increased mesocarp and kernel oil yields are the main targets in oil palm breeding. Mesocarp oil yield (OY) is improved by increasing the fresh fruit bunch (FFB) yield and the OTB ratio. FFB is directly measured by weighing all the bunches produced by the palm, while OTB is measured from three bunch components, namely, ratios of FTB, MTF, and oil-to-wet mesocarp (OTWM). These are the major elements determining the volume of palm oil, and they are the most important traits considered in individual palm selection for commercial seed production.
Significant QTLs for OY were detected on linkage groups 1, 6, and 2, which correspond to chromosomes 3, 7, and 8, respectively. However, two markers (SNPM00957 and SNPM01889) could not be placed specifically on chromosomes 3 and 8 as they fell on scaffolds that did not fit into the genome build. On LG1 (chromosome 3), genotype AA of SNPM00995 exhibits low OY compared to those exhibited genotypes AC and CC (Supplementary Table S4). As for marker SNPM01889 on LG2 (chromosome 8), palms with genotypes AG and GG exhibited higher OY than those showing AA. The GG genotype of marker SNPM05057 on chromosome 7 possessed significantly lower OY than those showing AA and AG genotypes. The oil palm planting materials are hybrid seeds known as tenera, produced from crossing between the dura and pisifera parental palms. In oil palm breeding program, oil yield is estimated mainly to determine the combining ability of the dura and pisifera parents. In addition, breeders also focus on measuring the subcomponents such as OTB, MTF, and OTWM. The OTB is influenced by another three bunch components (MFW, OTWM, and FTB). However, the markers linked to the QTLs for OY in these three chromosomes did not show any significant effect on the other element influencing OY. As such, it may not be feasible at this stage to justify the use of these markers in oil palm breeding and selection.
Nevertheless, gains in OY can actually be achieved by improving OTB (Soh 1999). We found significant QTLs associated with OTB on chromosomes 4 and 8. Individual palms with genotype GG had higher OTB than the alternative genotypes of marker SNPM02805 on chromosomes 4. On chromosome 8, palms exhibiting genotype TT of SNPM03231 gave higher OTB than those carrying the AA and AT genotypes. We also identified another QTL for OTB, SNPM00402, not very far from marker SNPM03231. Palms carrying genotype AA of SNPM00402 possessed higher OTB than palms with AG and GG genotypes.
MTF is another strong determinant of OY. QTLs significantly associated with MTF were detected on chromosomes 3, 4, 7, 11, and 15. Palms exhibiting the homozygous genotypes AA of SNPM02063 (chromosome 3), GG of SNPM02805 (chromosome 4), TT of SNPM00732 (chromosome 7), AA of SNPM01772 (chromosome 11), and GG of SNPM02015 (chromosome 15) had higher MTF (mean > 48.4%) than those with the other genotypes. Besides the QTL for MTF detected in this study, Billotte et al. (2010) and Jeennor and Volkaert (2014) also reported QTLs for MTF and OTF, respectively, on chromosome 3. Jeennor and Volkaert (2014) reported QTL for other bunch component, namely, FTB ratio, near the QTL for MTF identified in our study on chromosome 7 (Fig. 4). On chromosome 15, the QTL for MTF discovered in the current study is located near the QTL for MTF described previously (Billotte et al. 2010). On the same chromosome, Billotte et al. (2010) also reported another QTL for KTF, a trait negatively correlated with MTF. This can be an interesting region to search for candidate genes influencing MTF and KTF. The QTLs associated with MTF detected on chromosomes 4 and 11 appear to be specific to the Angola population.
In oil palm breeding, fruit size is measured by the MFW parameter. We found significant QTLs associated with MFW on chromosomes 4, 5, and 6. On chromosome 4, palms carrying genotype GG of SNPM02805 had higher MFW compared to those carrying GC and CC. Individual palms that possessed AA genotype of SNPM03826 on chromosome 5 had significantly higher MFW. The CC genotype of SNPM01664 from chromosome 6 exhibited higher MFW compared to the AA and AC genotypes. Correlation analysis showed that MFW is positively correlated with MTF, OTB, and OY (Table 2). In addition, previous studies have also shown that selection for high MFW may result in higher oil content in oil palm bunches (Rajanaidu and Jalani 1994; Sharma and Tan 1997). Selection for high MFW is expected to generate the largest absolute quantity of mesocarp, resulting in increased oil yield (Kushairi et al. 2003). Interestingly, QTLs associated with MFW were also reported by Billotte et al. (2010) and Jeennor and Volkaert (2014), located at the opposite end of chromosome 5. These groups also described QTLs for other bunch components such as MTF and OTDM on chromosome 6 which significantly associated with marker mEgCIR3826 using different mapping populations.
On chromosome 4, we found a QTL with pleiotropic effect, i.e., SNPM02805. The marker was associated with OTB, MFW, and MTF. Our results also indicate positive correlation among OTB, MTF, and MFW (Table 2). We observed similar effects of the genotypes on the three traits (Fig. 3). Palms with genotype GG had significantly higher MFW, OTB, and MTF compared to those with genotypes GC and CC (see Supplementary Table S4 and Fig. 3), suggesting that the same QTL is influencing the three bunch traits. Previous studies did not report QTLs linked to bunch component on chromosome 4. As such, the QTLs uncovered in this study maybe unique to the Angola germplasm. Similarly, the QTLs for OTB on chromosome 8 and QTLs for MTF on chromosome 11 described above were discovered only in this germplasm. These represent interesting loci for selection in breeding programs involving Angola germplasm.
Significant QTLs influencing vegetative traits, such as HI and RL, were detected on chromosomes 2, 4, 10, and 16, respectively. Palms with genotypes AA of SNPM00957 (chromosome 2) had lower HI than those with AT and TT. With regard to RL, palms that showed genotype CC of markers SNPM02255 (chromosome 2) and SNPM00204 (chromosome 4) and genotype TT of markers SNPM00736 (chromosome 10) and SNPM04445 (chromosome 16) revealed significantly shorter rachis than palms having other genotypes. Billotte et al. (2010) also reported QTLs associated with rachis length on chromosomes 2, 4, and 16. The QTLs for RL detected on chromosome 10 are unique for Angola population. Blast analysis of the flanking sequences of markers SNPM000957 and SNPM04445 did not show any significant association to known genes. Nevertheless, the findings suggest that it may be worthwhile to further investigate these genomic regions for genes influencing vegetative traits in oil palm.
Kernel oil earned ~ US$0.9 billion in 2015 for the Malaysian oil palm industry. The oil, which is rich in medium chain fatty acids, is mainly used in the oleo-chemical industry. Thus, increasing kernel yield can be profitable. Palms producing more kernels have been identified (Rajanaidu et al. 1996) and used in breeding for higher kernel yield. Here, we detected a QTL influencing KTB on chromosome 7. Individual palms with the TT genotype of marker SNPM02219 produced significantly lower KTB than palms with the CC and CT genotypes. The correlation was negative between KTB and OY as well as with the other oil-related components contributing to OY. As such, increasing the kernel yield will likely decrease the mesocarp oil yield, as also observed by Rajanaidu and Jalani (1994). This suggests that different planting materials need to be developed to produce more kernel or mesocarp oil. Development of alternative planting materials provides the oil palm industry an opportunity to diversify its products.
Highest number of QTLs associated with bunch components was observed on chromosome 5. These QTLs were mostly reported by other groups (Billotte et al. 2010; Jeennor and Volkaert 2014; Teh et al. 2016). Besides chromosome 5, QTLs for bunch components that are specific to advanced breeding populations were also observed on chromosomes 2 (Kwong et al. 2016) and 10 (Billotte et al. 2010; Jeennor and Volkaert 2014; Teh et al. 2016). As for the vegetative traits, QTLs specific to the advanced breeding populations were found on chromosome 6 (Billotte et al. 2010). Previous reports mostly utilized crosses or palms derived from advanced breeding lines (AVROS, Yangambi, La Me, Deli, Ulu Remis, Johor Labis, and Gunung Melayu) with limited genetic variability. As such, the above reports likely identified alleles already available in the main breeding program, which provide limited opportunity to improve diversity of the cultivated oil palms. Although there might be slight differences on the selection methods and criteria imposed on the populations, the ultimate objectives of oil palm breeding are similar across the world. Thus, the selection introduced may have resulted in preservation of similar sets of genes in these breeding populations.
For bunch components, QTLs common in both the Angola materials and advanced breeding populations were detected on chromosomes 3, 5, 6, 7, and 15. Similarly, common QTLs associated with vegetative traits were also found on chromosomes 2, 4, and 16. These QTLs may generally be applied for selection across a wider range of genetic background.
From the present work, Angola-specific QTLs for bunch components were found on chromosomes 4, 8, and 11. These QTLs revealed significant effects on their respective traits (MFW, MTF, OTB). We also detected a QTL significantly associated with RL on chromosome 10. These QTLs are potentially useful in introgression of desired regions from selected Angola palms to advanced breeding lines for improving the genetic diversity as well as bunch and oil yieldassociated traits.
In this study, we have extensively mapped QTLs associated with bunch components and vegetative parameters identified by other groups on to the genome build (Singh et al. 2013). This also allowed for comparison of genomic loci influencing these traits across various oil palm genetic backgrounds. Identifying QTLs close to the genomic regions reported previously for the same or associated traits adds confidence to the QTLs uncovered in this study, despite the fact that these QTLs explain a small proportion of trait variation. Search can be carried out for candidate genes in the regions influencing the selected traits across different genetic backgrounds. Expression analysis of these genes will provide a better understanding of their genetic control over the traits. In addition, new markers can be developed using the sequence of the candidate genes to further saturate the QTL regions. Such effort would also increase the confidence levels of the QTLs and lead to identification of genes responsible for the traits under consideration.
References
Billotte N et al (2010) QTL detection by multi-parent linkage mapping in oil palm (Elaeis guineensis Jacq.) Theor Appl Genet 120:1673–1687
Blaak G, Sparnaaij L, Menedez T (1963) Breeding and inheritance in the oil palm (Elaeis guineensis Jacq.) II. Methods of bunch quality analysis JW Afric Inst For Oil Palm Res 4:146-155
Corley RHV, Hardon JJ, Tan GY (1971) Analysis of growth of the oil palm (Elaeis guineensis Jacq.) I. Estimation of growth parameters and application in breeding. Euphytica 20:307–315
Doyle JJ (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Efron B, Tibshirani RJ (1994) An introduction to the bootstrap. CRC Press, Boca Raton
Fraley C, Raftery AE (2007) Model-based Methods of Classification: Using the mclust Software in Chemometrics. J Stat Softw 18(6):1–13
Friedman J, Hastie T, Tibshirani R (2010) Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33:1
Henderson CR (1975) Best linear unbiased estimation and prediction under a selection model. Biometrics:423–447
Jeennor S, Volkaert H (2014) Mapping of quantitative trait loci (QTLs) for oil yield using SSRs and gene-based markers in African oil palm (Elaeis guineensis Jacq). Tree Genet Genomes 10:1–14
Kennedy B, Quinton M, Van Arendonk J (1992) Estimation of effects of single genes on quantitative traits. J Anim Sci 70:2000–2012
Krzywinski M et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Kushairi A, Rajanaidu N, Mohd Din A, Isa Z, Noh A, Junaidah J (2003) Performance of Angola genetic materials. In: Proceedings of the Seminar on Progress of PS1 and PS2 Planting Materials and release of Elite Germplasm to Industry. pp 75–90
Kushairi A, Mohd Din A, Rajanaidu N (2011) Oil palm breeding and seed production. In: Mohd Basri W, Choo YM, Chan KW (Eds) Further advances in oil palm research (2000-2010) volume 1. Malaysian Palm Oil Board, Bangi, Selangor, pp 47–101
Kwong QB et al (2016) Development and validation of a high-density SNP genotyping array for African oil palm. Mol Plant 9:1132–1141
Lee M et al (2015) A consensus linkage map of oil palm and a major QTL for stem height. Sci Rep 5
Li J, Das K, Fu G, Li R, Wu R (2011) The Bayesian lasso for genome-wide association studies. Bioinformatics 27:516–523
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lockhart R, Taylor J, Tibshirani RJ, Tibshirani R (2014) A significance test for the lasso. Ann Stat 42:413
Meuwissen T, Hayes B, Goddard M (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Montoya C et al (2013) Quantitative trait loci (QTLs) analysis of palm oil fatty acid composition in an interspecific pseudo-backcross from Elaeis oleifera (HBK) Cortés and oil palm (Elaeis guineensis Jacq.) Tree Genet Genomes 9:1207–1225
Noh A, Rajanaidu N, Kushairi A, Mohd R, Mohd D, Mohd I, Saleh G (2002) Variability in fatty acids composition, iodine value and carotene content in the MPOB oil palm germplasm collection from Angola. J Oil Palm Res 14:18–23
Ong P, Maizura I, Abdullah N, Rafii M, Ooi L, Low E, Singh R (2015) Development of SNP markers and their application for genetic diversity analysis in the oil palm (Elaeis guineensis). Genet Mol Res 14:12205–12216
Pootakham W et al (2015) Genome-wide SNP discovery and identification of QTL associated with agronomic traits in oil palm using genotyping-by-sequencing (GBS). Genomics 105:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rajanaidu N, Jalani B (1994) Potential sources of lauric oils for the oleochemical industry. In: Proceedings of the World Conference and Exhibition on Lauric Oils: Sources, Processing and Applications AOCS, Manila, Philippines. pp 47–50
Rajanaidu N, Jalani S, Domingos M (1991) Collection of oil palm (Elaeis guineensis) germplasm in Angola Berita ISOPB 8:2–3
Rajanaidu NJ, Kushairi BS, Din AM, Rafill AM (1996) Breeding for high kernel planting material PORIM Series 3 (PS3)
Segura V, Vilhjálmsson BJ, Platt A, Korte A, Seren Ü, Long Q, Nordborg M (2012) An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat Genet 44:825–830
Sharma M, Tan Y (1997) Oil palm breeding programme and the performance of D×P planting materials at United Plantations Berhad. Planter 73:591–610
Singh R et al (2009) Mapping quantitative trait loci (QTLs) for fatty acid composition in an interspecific cross of oil palm. BMC Plant Biol 9:1
Singh R et al (2013) Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500:335–339
Soh A (1999) Breeding plans and selection methods in oil palm. In: Symposium on the science of oil palm breeding. Proceedings 1992 Montpellier, Francia, vol L-0435. PORIM
Teh CK, Ong AL, Kwong QB, Apparow S, Chew FT, Mayes S, Mohamed M, Appleton D, Kulaveerasingam H (2016) Genome-wide association study identifies three key loci for high mesocarp oil content in perennial crop oil palm. Sci Report 6:19075. https://doi.org/10.1038/srep19075
Tian F et al (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43:159–162
Tibshirani R (1996) Regression shrinkage and selection via the lasso. J R Stat Soc Ser B Methodol:267–288
Ting N-C et al (2014) High density SNP and SSR-based genetic maps of two independent oil palm hybrids. BMC Genomics 15:1
VanRaden P (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Waldmann P, Mészáros G, Gredler B, Fuerst C, Sölkner J (2013) Evaluation of the lasso and the elastic net in genome-wide association studies. Front Genet 4:270
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Zhang F, Guo X, Deng H-W (2011) Multilocus association testing of quantitative traits based on partial least-squares analysis. PLoS One 6:e16739
Zhou X, Stephens M (2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44:821–824
Acknowledgements
We acknowledge the Director General of MPOB for permission to publish the research findings. We thank the Breeding group for maintaining the Angola germplasm.
Funding
We also acknowledge MPOB’s support through the DNA Chip Program (R000999000-RB01-J) in funding the SNP genotyping and bioinformatics efforts. The project was also supported by the US National Science Foundation Collaborative Research Grant 473 DBI-1458515 to SX.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Data archiving statement
All data are available at figshare (https://figshare.com/s/183f196ff4dcf0303b1b).
Additional information
Communicated by W. Ratnam
Rights and permissions
About this article

Cite this article
Ithnin, M., Xu, Y., Marjuni, M. et al. Multiple locus genome-wide association studies for important economic traits of oil palm. Tree Genetics & Genomes 13, 103 (2017). https://doi.org/10.1007/s11295-017-1185-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1185-1