
Abstract
A fundamental goal of evolutionary biology is to understand how environment shapes genetic variation through its effect on demographic processes and through natural selection. In non-model species, transcriptome sequencing generates large single nucleotide polymorphism (SNP) panels to disentangle these influences. Quercus lobata (valley oak) offers an excellent system for such analyses because it has stably occupied a climatically heterogeneous landscape throughout California. We used 220,427 diallelic SNPs from 22 individuals identified against a recently assembled reference transcriptome to (1) quantify transcriptome-wide associations of SNPs with climate indicative of demographic responses to climate, (2) identify SNPs especially associated with climate and thus potential targets of natural selection, and (3) test the hypothesis that genetic diversity is high in climate-adaptive candidate genes. Constrained ordinations (redundancy analysis) and variance partitioning showed that genetic structure in Q. lobata was explained by spatial location (49 %) and climate (24 %), especially minimum temperature and summer/spring precipitation balance, suggesting that climate influences neutral demographic processes and gene flow. After accounting for underlying structure, individual-based environmental association analyses identified 79 SNPs from 49 transcripts as candidates under natural selection by climate. These candidate genes had significantly higher SNP rates per base pair per locus (θ W), nucleotide diversity (π), and gene diversity (G) than non-candidate genes. These results provide preliminary support for the hypothesis that balancing selection maintains diversity in climate-adaptive genes. Climate has likely shaped both population demography and local adaptation in valley oak.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The environment plays a fundamental role in shaping the geographic structure of genetic variation through its effect on demographic processes and through natural selection. In plant populations, climate can directly affect population expansion, contraction, and migration or alter flowering time and mating patterns (Cleland et al. 2007; Davis 1976). As a result of these “neutral” processes, genome-wide genetic variation is expected to associate with climate gradients (Eckert et al. 2010a; Sexton et al. 2014). In contrast, natural selection by climate would act largely on specific loci, such that associations with climate at particular loci should be independent of those with the background genetic structure (Eckert et al. 2010b; Hancock and Di Rienzo 2008; Keller et al. 2011). These genetic variants under selection along climate gradients within a species likely play a role in local adaptation and are of particular interest (Endler 1986). Because different alleles are favored in different environments (i.e., a form of balancing selection species wide), genetic variation at these loci is expected to be higher than the typical locus, which might be more likely to be under purifying selection (Lasky et al. 2014).
With genome-wide single nucleotide polymorphism (SNP) data sampled across environments, it is now possible to identify genetic variation under natural selection by local climate. One cost-effective way to identify large numbers of SNPs in non-model organisms is to perform whole-transcriptome sequencing (mRNA-Seq) from widely distributed samples and compare sequence variation, ignoring transcript abundance (Cánovas et al. 2010; Geraldes et al. 2011; Wang et al. 2009). An advantage of this approach is that it targets coding regions of the genome, which are commonly targets of selection and likely to underlie trait variation, and flanking untranslated regions, which play a role in gene expression (Barrett et al. 2012; Schork et al. 2013). Moreover, as genome size increases, enrichment for functional genomic regions can become important. Range-wide SNP data can then effectively be used in environmental association analyses to identify putatively adaptive genetic variation that displays exceptionally strong associations with the environment (De Mita et al. 2013) or in phenotypic association analyses to understand the genetic basis of trait variation (Atwell et al. 2010; Holliday et al. 2010; Neale and Savolainen 2004).
A number of powerful environmental association (“outlier”) analyses have been proposed that test whether specific loci are especially associated with a given environmental variable after accounting for background associations due to population structure (Coop et al. 2010; De Mita et al. 2013; Eckert et al. 2010b). For designs in which samples are scattered along the environmental gradient, rather than clustered in “populations,” linear mixed modeling approaches are an excellent choice (De Mita et al. 2013; Yoder et al. 2014). These models can effectively account for population structure using a kinship matrix of relatedness among individuals, are computationally efficient for large SNP data sets (Kang et al. 2008; Yu et al. 2006), and have low false-positive rates (Frichot et al. 2013), although they do not explicitly model population history (Günther and Coop 2013). One such method, EMMAX, was developed for genotype-phenotype associations and has been shown to outperform other similar approaches in accounting for underlying population structure (Kang et al. 2010; Sul and Eskin 2013). When applied to climate data, rather than phenotypic data, significant associations can be interpreted as candidate SNPs under natural selection by climate (Frichot et al. 2013; Yoder et al. 2014). Using the model in this way does not suggest that genotype causes climate, but rather it is a convenient statistical means of assessing the expected correlations among the variables of interest (Furlotte et al. 2011), and further, it can be assumed that adaptive phenotypes mediate those associations between SNP and climate (Eckert et al. 2010b). Accounting for population structure can minimize false-positive rates; however, all environmental association approaches still suffer from elevated false-negative rates due to some removal of true signal from the process of accounting for genetic structure. Environmental association analyses are especially powerful in highly outcrossing trees, such as oaks (Quercus), because linkage disequilibrium decays within a few hundred base pairs (Alberto et al. 2013; Brown et al. 2004; Kremer et al. 2012; Sork et al. 2016), meaning that significant associations are likely to be near the true target of selection (Neale and Savolainen 2004).
Our recently assembled transcriptome assembly and large SNP panel for Quercus lobata Née (valley oak) (Cokus et al. 2015) provides an excellent resource to disentangle how climate shapes underlying genome-wide genetic structure, presumably due to demographic processes (Gugger et al. 2013; Sork et al. 2010), versus the effect of selection by climate on specific SNPs (Sork et al. 2013). For comparison, several studies of the European oaks have identified candidate genes for climate-related traits such as timing of bud burst and response to drought stress based on differential gene expression experiments (Derory et al. 2006; Porth et al. 2005; Spieß et al. 2012). Some of these loci have been verified using nucleotide-based tests for signatures of natural selection (Derory et al. 2010) and other approaches (Alberto et al. 2013).
Q. lobata exhibits structured genetic diversity at the local to regional scales and has high potential for local adaptation relative to oaks in eastern North America and Europe, in part because its distribution has remained stable in a topographically complex area through recent glacial cycles unlike many oaks elsewhere (Grivet et al. 2006; Gugger et al. 2013). Q. lobata is currently threatened by land development and climate change (McLaughlin and Zavaleta 2012; Sork et al. 2010). Thus, it is especially important in Q. lobata to understand how climate shapes genetic variation through demographic processes, what genes are involved in adaptation to current environments, and how changing environments might impact adaptive genetic variation and population persistence for effective management.
Here, we use 220,427 diallelic SNPs previously identified in Q. lobata (Cokus et al. 2015) to (1) quantify transcriptome-wide associations of SNP variation with climate indicative of demographic responses to climate, (2) identify specific loci that are especially associated with climate and thus potential targets of natural selection, and (3) test the hypothesis that candidate genes for adaptation to climate gradients have higher genetic diversity than non-candidate genes.
Materials and methods
Sampling
Poly-A-purified mRNA libraries from 22 Q. lobata samples from throughout its natural distribution (Fig. 1) were previously sequenced for a de novo transcriptome assembly and SNP discovery project that included other California oaks (Cokus et al. 2015). This draft reference transcriptome includes a mixture of complete and partial gene models (generally with UTRs and introns), many of which contain Pfam protein domains (Finn et al. 2014; Jones et al. 2014), and a subset of which were found to be orthologous with Arabidopsis genes from The Arabidopsis Information Resource (TAIR) (Swarbreck et al. 2008). Inferred Gene Ontology (GO) (Ashburner et al. 2000) associations for numerous oak gene models were then available through TAIR and Pfam. From the total panel of over one million SNPs identified within and among the California oak species, we retained for the present study 220,427 diallelic SNPs that are variable within Q. lobata and for which genotype was not called in at most 2 of 22 samples (<10 %). For some analyses, we further restricted to those SNPs for which genotype was not called in at most one sample (193,428) or those with called genotype in all samples (155,465). Details on the SNP calling methods and quality control are in Cokus et al. (2015).

Distribution of valley oak (Quercus lobata) (blue) and sampling locations (black circles: small = one sample, large = two samples). Gray scale represents elevation (darker is higher)
Genetic structure and association with climate
To assess genetic structure and its association with spatial coordinates and climate variables, we performed redundancy analysis in vegan 2.0-7 (Oksanen et al. 2015) in R 3.0.0 using the completely called SNP set. Redundancy analysis is a multivariate analog of linear regression when there are multiple response variables (SNPs) and multiple explanatory variables (climate and space) and makes similar assumptions as principal component analysis (Legendre and Legendre 1998; ter Braak 1986). We further performed two partial redundancy analyses to partition the variance into the part explained uniquely by climate variables, that explained uniquely by spatial coordinates (e.g., due to spatial autocorrelation and phylogeographic structure), and the joint influence of these which could not be disentangled (Borcard et al. 1992; Økland 1999). The analysis was repeated for each type of SNP: synonymous, nonsynonymous, and noncoding. Statistical significances of the full models were tested via permutations. Redundancy analysis has been successfully applied to Arabidopsis and oaks to address similar questions about the roles of climate and spatial variables (Gugger et al. 2013; Lasky et al. 2012) and has been recommended over traditional Mantel and partial Mantel tests for its superior statistical properties (Legendre and Fortin 2010).
Derived climate data were drawn from a U.S. Geological Survey Spline Model designed for use in assessing plant-climate relationships (Rehfeldt 2006) (http://forest.moscowfsl.wsu.edu/climate/). We retained the five variables that are thought to be important determinants of Q. lobata’s distribution (McLaughlin and Zavaleta 2012; Sork et al. 2010) and that were not highly correlated with each other, including growing degree-days above 5 °C, mean maximum temperature of the warmest month, mean minimum temperature of the coldest month, growing season precipitation, and summer/spring precipitation balance. Spatial coordinates included latitude, longitude, squared terms, cubed terms, and cross products to account for nonlinear associations of genetic variation with spatial variables (Borcard et al. 1992).
SNPs under selection
To identify individual SNPs that are especially correlated (i.e., outliers) with any of the five climate variables, we performed linear mixed model correlations in EMMAX (intel-binary-20120210), as this effectively accounts for background genetic structure due to “hidden” relatedness or shared phylogeographic history via a pairwise “kinship matrix” among individuals based on the Balding-Nichols method (Balding and Nichols 1995; Kang et al. 2010; Sul and Eskin 2013). Standard linear mixed models as we implemented them have been evaluated against other individual-based environmental association methods, showing that they have very low false-positive rates and thus are conservative, and they have the benefit that no parameter optimization is necessary (Frichot et al. 2013). Although they can have high false-negative rates (i.e., low power), we prefer to be conservative, given our small sample size. For this analysis, we started with the 193,428 SNP set with at most one uncalled individual per locus. The kinship matrix was estimated based on a subset of 32,551 loci that were at least 500 bp apart when located on the same contig to ensure high likelihood of freedom from dependence mediated by linkage disequilibrium (and a version based on all SNPs was also tried, with similar results and hence omitted) (Alberto et al. 2013; Brown et al. 2004; Sork et al. 2016). Because redundancy analyses did not reveal any distinctions among types of SNPs (i.e., coding or noncoding; see “Results”), SNPs for the kinship matrix were randomly chosen with respect to type. The 70,639 SNPs with a minor allele frequency of at least 4 (≥10 %) of the total 2 × 22 = 44 alleles were tested for associations with climate variables. Consistent with common practice, this threshold serves as an additional filter to reduce the false positives due to potential high-leverage data points from rare alleles. Multiple testing was adjusted using the false discovery rate (Q) method of QVALUE 1.1 (Benjamini and Hochberg 1995; Storey and Tibshirani 2003). Finally, as a “validation” of the results for significant climate-associated SNPs from EMMAX, we performed partial Mantel tests of climate distance with genetic distance controlling for geographic distance. Relative to EMMAX, the partial Mantel tests essentially reverse the dependent and independent variables to be consistent with their hypothesized causal relationship. Geographic distance was calculated assuming a spherical model (WGS84) of the earth, climate and SNP allele frequency distance were calculated by Euclidean distance, and partial Mantel tests were performed in the vegan package in R.
Candidate genes from literature and functional annotations
We searched our data set for previously published climate-related candidate genes to determine whether SNPs from those genes were also among the top associations from EMMAX. We first examined our dataset for candidate genes reported elsewhere for oaks, specifically 213 drought and osmotic stress genes (Porth et al. 2005), 144 bud burst and flowering genes (Alberto et al. 2013; Derory et al. 2006) (http://www.evoltree.eu/), and 26 other climate-related genes (Sork et al. 2016). We searched for these genes in the reference transcriptome using USEARCH 7.0 (Edgar 2010) with thresholds of 92 % nucleotide identity and E value 10−10.
We also searched the Q. lobata transcriptome GO associations (Cokus et al. 2015) for keywords suggesting roles in responses to the tested climate variables. These included response to osmotic stress or homeostasis (GO IDs: 0006970, 0006972, 0009270, 0009992, 0030104, 0047484, 0071470) or water stimulus/deprivation (0009270, 0009414, 0009415, 0009819, 0042631, 0071462, 2000070), which might be related to growing season precipitation or summer/spring precipitation balance; response to heat (0034605, 0009408), heat acclimation (0010286, 0070370), or heat shock protein binding (0031072), which might be related to mean maximum temperature; response to cold or freezing (0050826, 0070417, 0009409) or cold acclimation (0009631), which might be related to mean minimum temperature; and flower/floral or leaf development or morphogenesis (0009908, 0009965, 0010093, 0010150, 0010338, 0010358, 0048366, 0048437, 0048438, 0048439, 0048444, 0048449, 0048464, 0048833), flower photoperiodism (0048573–0048575), or regulation of these processes (0009909, 0009910, 0009911, 0010080, 0048579, 0048577, 0048578, 0048586, 0048587, 0060860, 0060862, 2000024, 2000025, 2000028), which might be associated with a variety of climate variables, including growing degree-days, temperature, and precipitation (Hunter and Lechowicz 1992; Nizinski and Saugier 1988; Vitasse et al. 2011). We grouped flower and leaf development-related candidate genes together because flowers and leaves emerge from the same buds at almost the same time in Q. lobata.
We used hypergeometric tests (equivalent to one-tailed Fisher exact tests in this context) to determine whether SNPs from candidate genes from the literature and GO functional annotations were enriched in the top 5 % of associations from EMMAX with their respective climate variable. We also used one-sided Wilcoxon rank-sum tests to ask whether SNPs in those same candidate genes had stronger associations with their respective climate variables than non-candidate gene SNPs, as indicated by the P values from EMMAX.
Genetic diversity in candidate genes
We tested the hypothesis that genetic diversity is higher in climate-adaptive candidate genes than non-candidate genes using one-sided Wilcoxon rank-sum tests. We quantified genetic diversity with θ W or Watterson’s theta, which is a measure of SNP rate per bp per locus (Watterson 1975); π or nucleotide diversity, which is a measure of SNP rate per bp per locus weighted by frequency in the population (Begun et al. 2007; Nei and Li 1979); and G or Weir’s gene diversity, which is the average expected heterozygosity across all SNPs within a locus (Nei and Roychoudhury 1974; Weir 1996). These measures capture a range of concepts of genetic diversity from SNP rate irrespective of population allele frequency (θ W) to allele frequency in the population irrespective of SNP rate (G), and the combination of the two (π).
Genetic diversity measures were calculated based on the 220,427 SNP set with up to two uncalled genotypes per locus (as well as those with all genotypes called, but results were highly similar and thus omitted). We performed these tests separately for candidate genes from the literature, for those identified via GO associations, and for those identified in this study as top candidates associated with climate. For the former two tests, we restricted the non-candidate set to the 28,261 contigs with at least one gene model to avoid a downward bias of diversity estimates because many contigs without gene models are of low coverage and variant discovery power is reduced. As an additional control, we also did the same test omitting contigs with no SNPs. For the latter test, we restricted the non-candidate set to only the contigs that had SNPs that were tested in EMMAX because these were already a subset intentionally chosen to have at least a certain population allele frequency, which upwardly biases allele frequency-based diversity measures.
Results
Genetic structure and association with climate
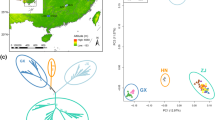
Samples from southern California sites (Malibu Creek State Park and Fort Tejon State Historic Park) are differentiated from other sites as shown by the redundancy analysis (Fig. 2a). Spatial and climate variables together are significantly associated with genetic variation (P < 0.005), and the variables most strongly associated with genetic structure are mean minimum temperature of the coldest month, summer/spring precipitation balance, growing degree-days, latitude, and longitude. In the partial redundancy analysis of climate with SNP variation controlling for spatial location, minimum temperature is most strongly associated with axis 1 and summer/spring precipitation balance is most associated with axis 2 (Fig. 2b). Climate variables explained 26 % of the total explainable genetic variance, spatial variables explained 53 %, and their joint effect explained the remaining 21 %. These results (not shown) are nearly identical whether considering all relevant SNPs or broken down by nonsynonymous, synonymous, and noncoding SNPs (Procrustes test: r > 0.98, P < 0.001).

a Full redundancy analysis model for association of climate and spatial variables with transcriptome SNPs. Black points display ordination based on genetic variation and represent the underlying genetic structure. The two isolated sets of points are the southern sites of Malibu Creek State Park (right) and Fort Tejon State Historic Park (bottom). Vectors give the direction and magnitude of association of climate and spatial variables with the genetic structure along redundancy axes (RDA) 1 and 2. b Partial redundancy analysis for association of climate variables with transcriptome SNP variation after partialling out the association of spatial variables with SNP variation. This represents the “pure” effect of climate on genetic structure and suggests that minimum temperature and summer/spring precipitation balance are the most important factors of those studied here in structuring transcriptome-wide SNP variation
SNPs under selection
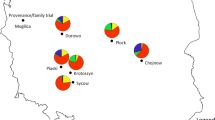
The strongest association of SNPs with any of the climate variables, after factoring out the underlying genetic structure via kinship, is with minimum temperature and growing season precipitation, and 12 of those SNPs from 10 contigs were statistically significant after adjustment for multiple testing (Q < 0.1) (Fig. 3 and Table 1). Given the large number of tests and small sample size, we also investigated the 67 other SNPs with associations of P < 0.0001, even if less than 500 bp apart. These include 10 SNPs from 6 contigs associated with growing degree-days, 20 additional SNPs from 12 contigs associated with growing season precipitation, 2 SNPs from 2 contigs associated with mean maximum temperature, 27 additional SNPs from 21 contigs associated with mean minimum temperature, and 7 SNPs from 6 contigs associated with summer/spring precipitation balance (Table 1). Of the resulting total of 79 distinct SNPs from 49 distinct contigs, 31 are nonsynonymous, 39 are synonymous, 2 are in 3′ untranslated regions (3′-UTRs), 3 were in 5′-UTRs, and 2 are undetermined. Nonsynonymous SNPs are not overrepresented in this list compared to the number in the overall sample of SNPs analyzed (hypergeometric test, P = 0.8). Of the 49 contigs, 4 are orthologous to A. thaliana genes, and 5 contained Pfam protein domains or TAIR annotations with GO annotations indicating their involvement in response to stimulus or stress (Table 1). All these climate-associated SNPs are also significantly associated in Mantel tests, providing a “validation” of the associations that considers the climate as the independent variable and genotype as the dependent variable (0.43 < r < 0.79, P < 0.008). Furthermore, QQ plots of the EMMAX results suggest that the false-positive rate is well controlled and thus the significant results are not likely spurious (Fig. S1 in Online Resource 1).

a Map showing the frequency of a particular nonsynonymous SNP (m01oak13412cC, nucleotide 2194) from a protein of unknown function (AT5G05190) significantly associated with growing season precipitation (background grayscale) (Q = 0.048, EMMAX). Large pie charts represent two valley oak individuals (four alleles) and small charts represent one individual (two alleles); they are colored by allele (blue = G, green = A). b Association of SNP frequency per individual with growing season precipitation for this same SNP (genotypes are 0 = A/A, 0.5 = A/G, 1 = G/G)
Candidate gene enrichment
Searches of the GO associations yielded 280 water-related genes, 128 heat-related genes, 127 cold-related genes, and 252 flower or leaf development-related genes. In addition, 298 of the 383 candidate genes from the oak literature (Alberto et al. 2013; Derory et al. 2006; Porth et al. 2005; Sork et al. 2016) are identifiable in our reference transcriptome and 233 of them have at least one SNP. Combining the candidate genes from GO with those from the oak literature, we observed from 27 to 122 candidate gene SNPs in the top 5 % of EMMAX associations with their respective climate variable (Table 4), but none have P < 0.0001 and thus none of these genes overlap with those identified as candidates in our EMMAX analyses. Specifically, SNPs from flower/leaf development-related genes are enriched in the top 5 % of EMMAX associations with growing degree-days, growing season precipitation, and minimum temperature (P < 0.003) (Table 4), but significant enrichment is not found for drought, heat, or cold gene SNPs in association with precipitation, maximum temperature, or minimum temperature, respectively (P > 0.26). Furthermore, Wilcoxon rank-sum tests show that SNPs from flower/leaf development-related genes have significantly lower P values for associations with growing degree-days and growing season precipitation compared to other genes (P < 0.037) and marginally significantly stronger associations with minimum temperature (P = 0.061) and summer/spring precipitation balance (P = 0.089) (Table 5). SNPs from cold genes also have stronger associations with minimum temperature (P = 0.012), and SNPs from heat genes have marginally significantly stronger associations with maximum temperature (P = 0.064).
Genetic diversity in candidate genes
Genetic diversity is higher in contigs containing SNPs that were from the top associations in EMMAX (i.e., those with P < 0.0001) compared to the other contigs tested in EMMAX, whether measured by θ W (P = 0.001), π (P = 0.00012), or G (P = 0.061) (Tables 1, 2, and 3). Genetic diversity is also higher for all measures in candidate genes from the oak literature and from the GO associations search when compared to all other contigs containing gene models (P < 2.2 × 10−16). However, this latter effect disappeared when only variable candidate genes (i.e., those containing SNPs) and variable non-candidate genes were considered (P > 0.132).
Discussion
Demographic response to climate
The transcriptome-wide genetic structure of oaks was shaped by climate. A substantial amount of SNP variation was explained by climate after controlling for spatial location (26 %), suggesting that climate shapes genomic variation independent of any association of climate with geographic location. Specifically, minimum temperature and summer/spring precipitation balance have the strongest association among those factors investigated. These associations likely reflect the effects of climate (especially minimum temperature) on demographic processes, such as population expansion, contraction, and establishment (James et al. 2011), and the influence of climate (especially temperature and precipitation balance) on gene flow through its influence on flowering phenology (Knight et al. 2005; Ortego et al. 2012).
Similar analyses of microsatellite variation in Q. lobata from 65 sample sites also support this role of minimum temperature and precipitation seasonality in shaping genetic variation and, in fact, suggest a potentially even larger role for climate than this study (Gugger et al. 2013; Sork et al. 2010). In the transcriptome data, the genetic distinction of two southern populations is pronounced (Fig. 2a), suggesting restricted gene flow among them and between other populations, possibly due to geographic barriers. Here, we do not observe the clear east–west structure (i.e., coast versus Sierra Nevada) that we did with microsatellites. Further, another study focusing on a subset of candidate genes from different localities also did not find east–west structure (Sork et al. 2016). However, the fact that three separate studies found genetic associations with similar climate variables provides strong evidence that environment is shaping genome-wide genetic structure through processes other than natural selection.
Natural selection by climate
Despite the genome-wide association with climate, we found strong evidence that natural selection by climate is important in local adaptation of valley oak. Even with a relatively small sample size of 22 individual trees, we identified 12 SNPs from 10 contigs significantly associated with climate variables after multiple testing adjustment (Q < 0.1) and an additional 67 SNPs from 39 contigs with very strong support (P < 0.0001) after factoring out background association of genetic structure with climate. Thirty-one of the 79 top SNPs are nonsynonymous and lead to amino acid substitutions, and 5 are in UTRs and thus could be involved in regulation of expression (Barrett et al. 2012); both of these types are consistent with functional significance.
A few of the SNPs are in genes with known roles in response to stimulus or stress (e.g., SAUR-like auxin-responsive protein family), cold shock protein binding (zinc knuckle family protein), light response or photosynthesis (e.g., cryptochrome 1), and trichome development (myosin family protein with Dil domain) (Table 1). The latter is especially interesting because trichomes are thought to be important in drought tolerance in plants (Karkkäinen et al. 2004).
Some of the SNPs with significant climate associations follow a north–south gradient orthogonal to the neutral genetic structure suggested in other studies based on microsatellite variation from more sites than this study (Gugger et al. 2013; Sork et al. 2010) (e.g., Fig. 3). Overall, these candidate SNPs showed especially strong correlations with growing season precipitation and minimum temperature, which are variables that also seem to be important shapers of the underlying genetic structure, presumably through their influence on demography and mating patterns. Given that Q. lobata occupies largely water-limited environments with frequent droughts, finding compelling evidence for natural selection by precipitation is not surprising.
The climate-associated SNPs from the EMMAX analyses also tend to have especially strong associations with the first axis in the partial redundancy analyses of climate with SNPs conditioned on spatial variables (Wilcoxon rank-sum test, P = 5 × 10−8), although only 1 of 79 SNPs was in the top 5 % of associations (m01oak05422cC, nucleotide 1044). This partial redundancy analysis could be considered another means of controlling population structure while testing for SNP-climate associations, and there is a growing interest in applying multivariate ordination approaches to identify specific loci of interest in environmental association tests (Sork et al. 2013). Although our data do not allow for a rigorous assessment of the ability of the redundancy analysis model to effectively account for population structure, and while a number of factors could lead to differences between these methods, it is encouraging that the SNPs identified in EMMAX also tend to be strongly associated in the partial redundancy analysis (Sork et al. 2016).
Finally, candidate genes for climate adaptation that were identified in other studies are among the top associations with climate in our analyses. These especially include flowering and leaf development genes associated with growing degree-days and growing season precipitation; cold genes with minimum temperature; and heat genes with maximum temperature (Tables 4 and 5), lending additional support to their role in climate adaptation in oaks.
Future studies will consider a larger sample of localities that will increase the ability to detect specific SNPs varying along climate gradients.
Genetic diversity in candidate genes
We find preliminary support for the hypothesis that climate-adaptive genes have elevated levels of genetic diversity. The most compelling evidence comes from the candidate genes identified here as the top associations with climate variables in EMMAX (Table 1). These genes had θ W and π over 50 % higher than other genes that were tested in EMMAX (Table 2). Moreover, G was 0.200 in candidate genes compared to 0.188 in non-candidates, providing support for the main hypothesis across a range of types of diversity measures. The elevated genetic diversity summarized across all top associations should be statistically robust (Lohmueller et al. 2013), despite the possibility of occasional false positives at any particular SNP or gene. On the other hand, our analysis does not control for the fact that loci with more SNPs were subjected to more tests in EMMAX, thus increasing the chance of finding a SNP significantly associated with climate. Choosing only one random SNP per locus led to too small a sampling of climate-associated SNPs to test for differences with the background.
Candidate genes from the oak literature as well as candidate genes identified by GO associations had higher diversity than non-candidates on average (Table 2). However, when only variable candidate and non-candidate genes from GO or the literature were compared, the difference was not significant (Table 3), suggesting that those candidate genes were more likely to be variable than non-candidate genes, but not more variable than other genes with variation. It is possible that the candidate genes from the literature and GO consist of many conserved genes involved in global responses to environmental perturbation that are not necessarily involved in local adaptation.
High diversity and significant associations with climate gradients are patterns that are consistent with balancing selection and/or disruptive selection maintaining diversity in climate-adaptive loci by favoring different alleles in different climate contexts. Although our study provides mixed support for elevated diversity, similar conclusions have been drawn for candidate genes for locally adaptive abiotic stress responses first identified using differential gene expression analyses in Arabidopsis (Lasky et al. 2014). Alternatively, soft selective sweeps that lead to increased adaptive allele frequencies in different parts of the distribution have been observed in Medicago (Yoder et al. 2014). Future studies with the ability to ascertain haplotypes will further clarify this possibility.
Conclusions
Climate has likely shaped both demographic and adaptive evolutionary processes in valley oak. Even with small sample sizes, we were able to disentangle candidate SNPs underlying climate adaptation from the background association of genomic variation with climate. As a result, we find some support that putatively climate-adaptive genes may have unusually high genetic variation, which we hypothesize is the result of natural selection leading to local adaptation that maintains diversity. Our study further highlights that large sequencing data sets and individual-based SNP analyses offer powerful means of identifying genes important in adaptation and the overall influence of climate on the genome.
References
Alberto FJ, Derory J, Boury C, Frigerio J-M, Zimmermann NE, Kremer A (2013) Imprints of natural selection along environmental gradients in phenology-related genes of Quercus petraea. Genetics 195:495–512. doi:10.1534/genetics.113.153783
Ashburner M et al (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29
Atwell S et al (2010) Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 465:627–631. doi:10.1038/nature08800
Balding D, Nichols R (1995) A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica 96:3–12. doi:10.1007/bf01441146
Barrett LW, Fletcher S, Wilton SD (2012) Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci 69:3613–3634. doi:10.1007/s00018-012-0990-9
Begun DJ et al (2007) Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. Plos Biol 5:e310. doi:10.1371/journal.pbio.0050310
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300. doi:10.2307/2346101
Borcard D, Legendre P, Drapeau P (1992) Partialling out the spatial component of ecological variation. Ecology 73:1045–1055. doi:10.2307/1940179
Brown GR, Gill GP, Kuntz RJ, Langley CH, Neale DB (2004) Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc Natl Acad Sci U S A 101:15255–15260. doi:10.1073/pnas.0404231101
Cánovas A, Rincon G, Islas-Trejo A, Wickramasinghe S, Medrano J (2010) SNP discovery in the bovine milk transcriptome using RNA-Seq technology. Mamm Genome 21:592–598. doi:10.1007/s00335-010-9297-z
Cleland EE, Chuine I, Menzel A, Mooney HA, Schwartz MD (2007) Shifting plant phenology in response to global change. Trends Ecol Evol 22:357–365. doi:10.1016/J.Tree.2007.04.003
Cokus SJ, Gugger PF, Sork VL (2015) Evolutionary insights from de novo transcriptome assembly and SNP discovery in California white oaks. BMC Genomics 16:552
Coop G, Witonsky D, Di Rienzo A, Pritchard JK (2010) Using environmental correlations to identify loci underlying local adaptation. Genetics 185:1411–1423. doi:10.1534/genetics.110.114819
Davis MB (1976) Pleistocene biogeography of temperate deciduous forests. Geosci Man 13:13–26
De Mita S, Thuillet A-C, Gay L, Ahmadi N, Manel S, Ronfort J, Vigouroux Y (2013) Detecting selection along environmental gradients: analysis of eight methods and their effectiveness for outbreeding and selfing populations. Mol Ecol 22:1383–1399. doi:10.1111/mec.12182
Derory J et al (2006) Transcriptome analysis of bud burst in sessile oak (Quercus petraea). New Phytol 170:723–738. doi:10.1111/J.1469-8137.2006.01721.X
Derory J et al (2010) Contrasting relationships between the diversity of candidate genes and variation of bud burst in natural and segregating populations of European oaks. Heredity 104:438–448. doi:10.1038/Hdy.2009.134
Eckert AJ, Bower AD, González-Martínez SC, Wegrzyn JL, Coop G, Neale DB (2010a) Back to nature: ecological genomics of loblolly pine (Pinus taeda, Pinaceae). Mol Ecol 19:3789–3805. doi:10.1111/j.1365-294X.2010.04698.x
Eckert AJ, van Heerwaarden J, Wegrzyn JL, Nelson CD, R-I J, González-Martínez SC, Neale DB (2010b) Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., Pinaceae). Genetics 185:969–982. doi:10.1534/genetics.110.115543
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi:10.1093/bioinformatics/btq461
Endler JA (1986) Natural selection in the wild. Princeton University Press, Princeton
Finn RD et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi:10.1093/nar/gkt1223
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30:1687–1699. doi:10.1093/molbev/mst063
Furlotte NA, Kang HM, Ye C, Eskin E (2011) Mixed-model coexpression: calculating gene coexpression while accounting for expression heterogeneity. Bioinformatics 27:i288–i294. doi:10.1093/bioinformatics/btr221
Geraldes A et al (2011) SNP discovery in black cottonwood (Populus trichocarpa) by population transcriptome resequencing. Mol Ecol Resour 11:81–92. doi:10.1111/j.1755-0998.2010.02960.x
Grivet D, Deguilloux M-F, Petit RJ, Sork VL (2006) Contrasting patterns of historical colonization in white oaks (Quercus spp.) in California and Europe. Mol Ecol 15:4085–4093
Gugger PF, Ikegami M, Sork VL (2013) Influence of late quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol 22:3598–3612. doi:10.1111/mec.12317
Günther T, Coop G (2013) Robust identification of local adaptation from allele frequencies. Genetics 195:205–220. doi:10.1534/genetics.113.152462
Hancock AM, Di Rienzo A (2008) Detecting the genetic signature of natural selection in human populations: models, methods, and data. Annu Rev Anthropol 37:197–217
Holliday JA, Ritland K, Aitken SN (2010) Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis). New Phytol 188:501–514. doi:10.1111/j.1469-8137.2010.03380.x
Hunter AF, Lechowicz MJ (1992) Predicting the timing of budburst in temperate trees. J Appl Ecol 29:597–604
James PMA, Coltman DW, Murray BW, Hamelin RC, Sperling FAH (2011) Spatial genetic structure of a symbiotic beetle-fungal system: toward multi-taxa integrated landscape genetics. PLoS One 6:e25359. doi:10.1371/journal.pone.0025359
Jones P et al (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi:10.1093/bioinformatics/btu031
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723. doi:10.1534/genetics.107.080101
Kang HM et al (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354
Karkkäinen K, Løe G, ÅGren J (2004) Population structure in Arabidopsis lyrata: evidence for divergent selection on trichome production. Evolution 58:2831–2836. doi:10.1111/j.0014-3820.2004.tb01634.x
Keller SR, Levsen N, Ingvarsson PK, Olson MS, Tiffin P (2011) Local selection across a latitudinal gradient shapes nucleotide diversity in balsam poplar Populus balsamifera L. Genetics. doi:10.1534/genetics.1111.128041
Knight TM et al (2005) Pollen limitation of plant reproduction: pattern and process. Annu Rev Ecol Evol Syst 36:467–497. doi:10.1146/annurev.ecolsys.36.102403.115320
Kremer A et al (2012) Genomics of Fagaceae. Tree Genetics Genomes 8:583–610. doi:10.1007/s11295-012-0498-3
Lasky JR, Des Marais DL, McKay JK, Richards JH, Juenger TE, Keitt TH (2012) Characterizing genomic variation of Arabidopsis thaliana: the roles of geography and climate. Mol Ecol 21:5512–5529. doi:10.1111/j.1365-294X.2012.05709.x
Lasky JR et al (2014) Natural variation in abiotic stress responsive gene expression and local adaptation to climate in Arabidopsis thaliana. Mol Biol Evol 31:2283–2296. doi:10.1093/molbev/msu170
Legendre P, Fortin M-J (2010) Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Mol Ecol Resour 10:831–844. doi:10.1111/j.1755-0998.2010.02866.x
Legendre P, Legendre L (1998) Numerical ecology, 2nd edn. Elsevier, Amsterdam
Lohmueller Kirk E et al (2013) Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet 93:1072–1086. doi:10.1016/j.ajhg.2013.11.005
McLaughlin BC, Zavaleta ES (2012) Predicting species responses to climate change: demography and climate microrefugia in California valley oak (Quercus lobata). Glob Chang Biol 18:2301–2312. doi:10.1111/j.1365-2486.2011.02630.x
Neale DB, Savolainen O (2004) Association genetics of complex traits in conifers. Trends Plant Sci 9:325–330. doi:10.1016/J.Plants.2004.05.006
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A 76:5269–5273
Nei M, Roychoudhury AK (1974) Sample variances of heterozygosity and genetic diversity. Genetics 76:379–390
Nizinski JJ, Saugier B (1988) A model of leaf budding and development for a mature Quercus forest. J Appl Ecol 25:643–652
Økland RH (1999) On the variation explained by ordination and constrained ordination axes. J Veg Sci 10:131–136
Oksanen J et al. (2015) vegan: Community Ecology Package, 2.3 edn.
Ortego J, Riordan EC, Gugger PF, Sork VL (2012) Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol 21:3210–3223. doi:10.1111/j.1365-294X.2012.05591.x
Porth I, Koch M, Berenyi M, Burg A, Burg K (2005) Identification of adaptation-specific differences in mRNA expression of sessile and pedunculate oak based on osmotic-stress-induced genes. Tree Physiol 25:1317–1329
Rehfeldt G (2006) A spline model of climate for the western United States. USDA Forest Service, Port Collins
Schork AJ et al (2013) All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet 9:e1003449. doi:10.1371/journal.pgen.1003449
Sexton JP, Hangartner SB, Hoffmann AA (2014) Genetic isolation by environment or distance: which pattern of gene flow is most common? Evolution 68:1–15. doi:10.1111/evo.12258
Sork VL, Davis FW, Westfall R, Flint A, Ikegami M, Wang H, Grivet D (2010) Gene movement and genetic association with regional climate gradients in California valley oak (Quercus lobata Née) in the face of climate change. Mol Ecol 19:3806–3823. doi:10.1111/j.1365-294X.2010.04726.x
Sork VL, Aitken SN, Dyer RJ, Eckert AJ, Legendre P, Neale DB (2013) Putting the landscape into the genomics of trees: approaches for understanding local adaptation and population responses to changing climate. Tree Genetics Genomes 9:901–911. doi:10.1007/s11295-013-0596-x
Sork VL, Squire KC, Gugger PF, Levy E, Steele S, Eckert AJ (2016) Landscape genomic analysis of candidate genes for climate adaptation in a California endemic oak, Quercus lobata Née (Fagaceae). Am J Bot 103:1–13
Spieß N et al (2012) Ecophysiological and transcriptomic responses of oak (Quercus robur) to long-term drought exposure and rewatering. Environ Exp Bot 77:117–126. doi:10.1016/j.envexpbot.2011.11.010
Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445
Sul JH, Eskin E (2013) Mixed models can correct for population structure for genomic regions under selection. Nat Rev Genet 14:300–300
Swarbreck D et al (2008) The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36:D1009–D1014. doi:10.1093/nar/gkm965
ter Braak CJF (1986) Canonical correspondence analysis: a new eigenvector technique for multivariate direct gradient analysis. Ecology 67:1167–1179. doi:10.2307/1938672
Vitasse Y, François C, Delpierre N, Dufrêne E, Kremer A, Chuine I, Delzon S (2011) Assessing the effects of climate change on the phenology of European temperate trees. Agric For Meteorol 151:969–980. doi:10.1016/j.agrformet.2011.03.003
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi:10.1038/nrg2484
Watterson GA (1975) Number of segregating sites in genetic models without recombination. Theor Popul Biol 7:256–276
Weir BS (1996) Genetic data analysis II: methods for discrete population genetic data. Sinauer Associates, Sunderland
Yoder JB, Stanton-Geddes J, Zhou P, Briskine R, Young ND, Tiffin P (2014) Genomic signature of adaptation to climate in Medicago truncatula. Genetics 196:1263–1275. doi:10.1534/genetics.113.159319
Yu JM et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Acknowledgments
We thank E. Eskin, K. Lohmueller, M. Pellegrini, and A. Platt for helpful discussion. This project was funded by seed money from UCLA to VLS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Kremer
Data archiving statement
Illumina RNA sequence reads and SNP data are available through NCBI project accession PRJNA282155 and http://genomes.mcdb.ucla.edu/OakTSA/, following Cokus et al. (2015).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 15 kb)
Rights and permissions
About this article

Cite this article
Gugger, P.F., Cokus, S.J. & Sork, V.L. Association of transcriptome-wide sequence variation with climate gradients in valley oak (Quercus lobata). Tree Genetics & Genomes 12, 15 (2016). https://doi.org/10.1007/s11295-016-0975-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-016-0975-1