
Abstract
Cyanide is a nitrile which is used extensively in many industries like jewelry, mining, electroplating, plastics, dyes, paints, pharmaceuticals, food processing, and coal coking. Cyanides pose a serious health hazard due to their high affinity towards metals and cause malfunction of cellular respiration by inhibition of cytochrome c oxidase. This inhibition ultimately leads to histotoxic hypoxia, increased acidosis, reduced the functioning of the central nervous system and myocardial activity. Different physicochemical processes including oxidation by hydrogen peroxide, alkaline chlorination, and ozonization have been used to reduce cyanide waste from the environment. Microbial cyanide degradation which is considered as one the most successful techniques is used to take place through different biochemical/metabolic pathways involving reductive, oxidative, hydrolytic or substitution/transfer reactions. Groups of enzymes involved in microbial degradation are cyanidase, cyanide hydratase, formamidase, nitrilase, nitrile hydratase, cyanide dioxygenase, cyanide monooxygenase, cyanase and nitrogenase. In the future, more advancement of omics technologies and protein engineering will help us to recoup the environment from cyanide effluent. In this review, we have discussed the origin and environmental distribution of cyanide waste along with different bioremediation pathways and enzymes involved therein.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyanides are C≡N group containing compounds available in the form of nitriles, carbonitriles and cyanides. These compounds are extensively used in precious metal extraction industries and in agrochemical industries. These compounds are not only highly toxic to living beings but also recalcitrance to biodegradation (Luque-Almagro et al. 2011). Apart from being generated as industrial waste, cyanide wastes are also generated by degradation of cyanoglycosides and cyanolipids present in plants during raw plant processing. They are also released by microorganisms during their metabolic activity (Mekuto et al. 2016). Plants, fungi as well as bacteria are also synthesized cyanide through the process of cyanogenesis (Raquel et al. 2008; Bhalla et al. 2018). Physical and chemical methods of cyanide treatment are relied on uses of hazardous reagents and these processes are very expensive. Despite cyanide toxicity, few microorganisms are able to detoxify cyanides by using various enzymes present in their system. During the last few decades bioremediation is considered as one of the best alternatives to chemical treatment processes. Although bioremediation is an ecologically viable technique for cyanide degradation, their use at the industrial scale is still skeptical and under investigation at bench and pilot scale. The recent development of “omics” technology has contributed significantly in cyanide remediation process by gathering information in genome level from different microorganisms and converted them in genomic-proteomic-metabolomic techniques, called “cyanomics” (Luque-Almagro et al. 2016). In this review, we have discussed the origin and distribution of cyanide waste in the environment along with different bioremediation techniques adopted in the last few decades for curbing down the cyanide pollution. A detail enzymatic degradation pathway is also discussed.
Sources and distribution of cyanide in the environment
Cyanogenic compounds are ubiquitous in nature and produced by bacteria, fungi, plants and some insects during their various activities such as, at the time when they need to control mating behavior and need to fight against predators (Kumar et al. 2013; Frapolli et al. 2012; Baxter and Cummings 2006).
Around 2500 plant species were reported to have this kind of secondary metabolites in their system (Panter 2018). Cyanic/cyanide compounds are synthesized by many plants in the form of cyanolipids and cyanoglycosides which act as a deterrent against pests like insects and animals (Møller 2010). Ingestion of these plants causes acute or chronic plant poisoning in animals and humans. Pits and seeds of some plants (bitter almonds, apples, apricot, peaches, barley, lima beans, sorghum, flax seed) have a substantial amount of cyanoglycoside which can be further metabolized to produce cyanide; however, edible portion of plants have a low concentration of such compounds. Some species of bamboo shoots are bitter in taste and contains a cyanoglycoside known as taxiphyllin (Young 1954). Plants naturally synthesize these cyanogenic compounds using enzyme hydroxynitrile lyase. These compounds are in inactive forms and stored in one part of the cell and the activating enzyme is present in another part of the cell. Upon tissue damage due to pest/herbivore chewing activity or mechanical injury, the cyanogenic compounds and activating enzymes come into contact with each other and cyanide is then cleaved from the sugar, hence acting as a defense agent against pest and herbivore (Sharma et al. 2005).
To defy plants cyanide defense system, insects feeding on them slowly developed tolerance towards these chemicals by the process of co-evolution. These insects such as burnet moths e.g. Zygaena filipendulae not only became tolerant to cyanoglycosides but also started forming similar compounds like their host and also sequestered them from the host and utilized these cyanoglycosides against their predators. Zagrobelny et al. (2009) have identified the genes for cyanoglycoside synthesis in the six-spotted burnet moth using pyrosequencing. These cynaoglycosides not only act as a defense agent but also act as a pheromone. Female burnet discharges a trail of hydrogen cyanide which acts as an attractant for the male burnet moth. The male having a higher concentration of cyanide is preferred over others and during mating these cyanoglycosides are exchanged from male to female (Zagrobelny et al. 2007). These compounds provide an extra defense to eggs. They also act as a stored source of reduced sugar and nitrogen for the burnet moth.
Other than natural sources, cyanide compounds are also added to the environment by many anthropogenic activities. Cyanide compounds have many applications in the pharmaceutical industry, polymer manufacturing, mining and steel manufacturing, electroplating and agrochemical production (Kumar et al. 2013). The effluents from such industries are recalcitrant to treatment and pose many environmental problems. Agricultural practices (while using nitrile herbicides) also significantly contribute to increased cyanide level in soil and environment. Dichlobenil, ioxynil, bromoxynil and chlorothalonil are few nitrile based herbicides which are used as pest control agents in a variety of crops e.g. wheat, rice, corn, and barley (Lovecka et al. 2015). Cyanide is used as a laxative in precious metal mining as they form tight complexes with gold, silver, iron, heavy metals and also help them to solubilize from the ore. Cyanide is used in relatively dilute concentration for gold metal extraction too, known as the gold cyanidation process (He and Kappler 2017). Other than waste, road salt additives also contribute cyanide in the soil. During road construction, sodium ferrocyanide and ferric ferrocyanide (road deicing salt) are used as anti caking compounds. Due to sunlight exposure and microbial activity, ferrocyanide chemically degrade and cyanide ions are released into the soil (Paschka et al. 1999). In soil, the cyanide compounds are fairly mobile and move from soil to water and air. However, in the soil, the concentration of cyanide does not go up, mainly due to volatilization and microbial action (Paschka et al. 1999; ATSDR 2016).
Different strategies adopted in the last few decades for cyanide removal
Owing to their potential environmental toxicity, different cyanide detoxification strategies were adopted and modified during the last few decades. Many chemical and physical methods have been found useful in this detoxification process but numerous factors such as the chemical composition of the waste, its volume, the effluent quality, reagents availability are negatively affecting the feasibility of these methods (Akcil and Mudder 2003; Botz et al. 2015). In the last two decades, many conventional methods including natural, physical, chemical and biological methods (phytoremediation and microbial remediation) have been followed to remove cyanide containing waste from different environmental compartments. In the following section, we have discussed the cyanide bioremediation methods followed by the enzymatic degradation pathways.
Biological methods of cyanide removal
It is well evidenced that phytoremediation of thiocyanate and metal–cyanide complexes and microbial biodegradation of cyanide from ore mining wastewaters are the most successful methods of cyanide removal (Dash et al. 2009). In bacterial bioremediation process, free cyanide and metal-complexed cyanides are transformed to ammonia and bicarbonate; the free metals thus formed are either precipitated out from the solution or get adsorbed within the biofilm. In this context, it is worth mentioning that iron cyanides are less readily biodegraded and bioadsorbed than cyanide complexes of zinc, nickel, and copper (Akcil 2003; Botz 2001). Both aerobic and anaerobic biological treatments are followed to remove cyanide and thiocyanate after standardizing the environmental factors (temperature, pH, oxygen levels, and nutrition). For aerobic biological treatment of cyanide and thiocyanate many processes have been reported which include packed beds, rotating biological contactors (Campos et al. 2006), sequencing batch reactors, biological filters (White and Schnabel 1998), facultative lagoons, and activated sludge (Kaewkannetra et al. 2009).
Phytoremediation
Phytoremediation is one of the successful bioremediation strategies for cyanide detoxification. In this context Yu et al. (2004) have done substantial work on this method whereas a concise review on this topic was published by Kumar et al. (2017). Other plant species which were able to remove cyanide from soil and solutions are also listed in Table 1 (excluding the list published by Yu et al. 2004). Most of the plant species reported for phytoremediation were shown to have low tolerance and slow cyanide degradation rate. Phytoremediation is an extremely slow process which requires huge land area and cyanide concentration < 10 mg CN− L−. This process is therefore practically not feasible to implement in arid regions. While cyanogenic plants were reported to synthesize cyanoglycosides and cyanolipids, some non cyanogenic species (maize and some Chinese vegetation) were also reported to have cyanide degrading activity (Yu et al. 2004; Yu and Gu 2007). In contrast to microbial degradation, phytoremediation process was found superior in case of metal cyanide complexes (e.g. iron cyanide) (Aronstein et al. 1994). In this case, the plant species were not only tolerant but also showed positive growth pattern in the presence of cyanides. For example, Salix babylonica exhibited modified enzyme activities (superoxide dismutase, catalase, peroxidase), high rate of transpiration, increased chlorophyll content and soluble protein while engaged in phytoremediation of cyanide (Yu and Gu 2009). However, the fundamental biological mechanisms involved in this process are still unknown. A simplistic mechanism is discussed in the following section to understand the process.
The plant–microbe interactions (in symbiotic association) in rhizosphere emit many secondary metabolites and in some cases exuded biosurfactants (e.g., rhamnolipids). Biosurfactants help to increase the solubility as well as bioavailibilty of the hydrophobic organic nitriles (Volkering et al. 1998; Siciliano and Germida 1998). Beside this, phytosiderophores (present in roots) have also influenced sequestration and translocation of the metallocyanide in the plant (Taiz and Zeiger 2002). Sometimes fungi, when engaged in symbiotic association with plants, are able to increase root surface area and can detoxify toxic cyanide through stabilization, extraction or by degradation (Ebbs 2004). The root uptake of the organic pollutant is not easy as there is no transporter in the plasma membrane owing to the xenobiotic nature of the pollutant. The suberine layer of plant root endodermis acts as impermeable membrane and resists the toxic material flow directly into xylem by root apoplast (Taiz and Zeiger 2002). Translocation of the pollutants into the plant roots and within plant tissue is governed by simple diffusion mechanism which further depends on the hydrophobicity of the compounds (Trapp and McFarlane 1995). Once entered inside the tissue, these compounds are bound to chelators (GSH (γ-glu-cys-gly)/PCs- phytochelatins) and subsequently transported actively to the vacuole by ABC-type transporter (Pickering et al. 2000; Cobbett and Goldsbrough 2000; Mäser et al. 2001). In vacuoles, these conjugated compounds form more complex structure after reacting with sulfide molecules; after that these complex molecules can either stay in accumulated form within the plant tissue or may be metabolized. These stored complexes are further catabolized partially into stable intermediates (McCutcheon and Schnoor 2003) by the action of enzymes coming from endophytic microorganisms (Barac et al. 2004). These enzymes may modify these complexes by adding side groups which further increases solubility of these organic effluents and hence can initiate its degradation by phytoenzymes. The major enzymes involved in this process are β-cyanoalanine synthase, rhodanese, formamide hydrolyase, cyanide dihydratase (Miller and Conn 1980).
Among these enzymes, β-cyanoalanine synthase (CAS) is a ubiquitous enzyme and is able to catalyze the reaction between cysteine and cyanide which ultimately results in β-cyanoalanine and sulfide formation (Maruyama et al. 2001). β-cyanoalanine is a wide spread amino acid found in the plant kingdom. They are neurotoxic in nature and acted as a defense agent against herbivory (Piotrowski et al. 2001). It was also found that asparagines (nitrogen storage form) could be formed from β-cyanoalanine by the action of β-cyanoalanine hydrolase (Castric et al. 1972). In plants, rhodanese and formamide hydrolase are not very common enzymes (Miller and Conn 1980). Rhodanese has been reported in a few plants e.g. Brassica oleracea (Tomati et al. 1972), cassava (Emmanuel and Emmanuel 1981), Sorghum sp. (Myers and Fry 1978). Formamide hydrolase has been reported in Japanese apricot and loquat (Shirai 1978; Miller and Conn 1980). Inorganic cyanide is first transformed into formamide by formamide hydrolase which further can form formaldoxime and finally converted into formic acid and ammonia (Shirai 1978). The role of cyanide hydratases and cyanide dihydratases is discussed in a later section.
Microbial remediation
In microbial remediation process bacteria, fungi, algae and yeasts are involved in cyanide remediation. Temperature and pH are important parameters for determining the biodegradation rate and it was found that 20–40 °C temperature and 6–9 pH are optimum for microbial bioremediation process. Many fungal species such as Fusarium solani, Fusarium oxysporum, Penicillin miczynski, Scytalidium thermophilum, Trichoderma polysporum (either individually or in consortia) as well as many bacterial species belonging to the genera Pseudomonas, Bacillus, Rhodococcus and Serretia have been reported for their cyanide degrading ability (Barclay et al. 1998; Acera et al. 2017; Singh et al. 2018; Manso Cobos et al. 2015; Ibáñez et al. 2017; Maniyam et al. 2013; Bhalla et al. 2018). Varieties of enzymatic pathways are involved in the biotransformation and biodegradation processes which include degradative pathways (e.g. hydrolytic, oxidative, and reductive pathway forming NH3, HCO2H, CO2, CH4 and RCO2H) and assimilative pathways (substitution/transfer/synthesis). Most of the cyanide degrading microbes are aerobes; however anaerobic degradation of cyanide has also been reported and recently reviewed by Luque-Almagro et al. (2018). It was found that among all the degradative pathways, only reductive/hydrolytic pathways are feasible under anaerobic conditions (Fallon 1992). Enzymatic cyanide detoxification and cyanide assimilation by different microorganisms have been reviewed extensively and documented well in a number of reviews (Akcil and Mudder 2003; Samiotakis and Ebbs 2004; Baxter and Cummings 2006; Huertas et al. 2006; Dash et al. 2009; Gupta et al. 2010; Kumar et al. 2017; Luque-Almagro et al. 2016; Mekuto et al. 2016; Park et al. 2017). The following section has elaborately described the enzymatic degradation pathways of cyanide bioremediation.
Enzymatic degradation pathways of cyanides
Microbes employ different pathways to deal with toxic chemicals that are present in their habitat. The cyanide metabolizing microbes have followed five different pathways to mineralize the cyano compounds (Dash et al. 2009; Ebbs 2004). Table 2 described the various enzymes (along with their sources) involved in the enzymatic degradation pathway of cyanide biodegradation. In the following sections, the enzyme and their mechanism of action are discussed in detail.
Oxidative pathway
In the oxidative pathway the cyanide compounds are converted into ammonia and carbon dioxide using three different enzymes, viz., cyanase, cyanide monooxygenase, cyanide dioxygenase enzymes (Dash et al. 2009; Ebbs 2004).
Cyanase (EC 4.2.1.104)
Cyanase belongs to the lyase family of enzymes and generally inducible in nature. These enzymes have been reported in plants (Aichi et al. 1998), in cyanobacteria (Harano et al. 1997) and in many microorganisms. It is used by many organisms for cyanate detoxification when the concentration of cyanate is low in the system (Luque-Almagro et al. 2011). Cyanate and isocyanate (i.e. present in equilibrium) irreversibly carbomylated lysine into homocitrulline, modified the protein structure and further rendering them in the inactive or dysfunctional state (Koeth et al. 2013). Cyanate acts as a competitive inhibitor of nitrate and also oxidizes the reductively inactive enzymes and reactivates them. Cyanase catalyses the conversion of cyanate into carbon dioxide and ammonia in two steps processes. In the first reaction, the bicarbonate which acts as nucleophile reacts with cyanate (which is produced by its oxidation in soil) and forms an unstable carbamate as an intermediate which further spontaneously breaks down to form ammonia and carbon dioxide by decarboxylation reaction (Luque-Almagro et al. 2011). Earlier in their study, Luque-Almagro et al. (2008) reported an alkaliphilic bacterium Pseudomonas pseudoalcaligenes CECT5344 which showed good growth in different cyano compounds by using these compounds as a sole source of nitrogen. The microbe harbored two independent pathways for cyanide degradation. One pathway was cyanase mediated, consisting of cynFABDS gene cluster and another one was Nitrilase (NitC gene) enzyme mediated pathway. A recent article published by Cabello et al. (2018), reviewed the application of transcriptomics and proteomics techniques for getting insight into cyanide degradation pathway of Pseudomonas pseudoalcaligenes.
An earlier report published by Sung and Fuchs 1992, demonstrated that E. coli strain K-12 possessed cyanide degrading genes in the form of cyn operon. The regulatory protein known as CynR was usually present in the upstream of cyn operon. The molecular weight of CynR was 32-kDa and it belongs to LysR regulatory proteins (Sung and Fuchs 1992). The cynR promoter shared the overlapping region with cyn operon promoter on the opposite strand (Lamblin and Fuchs 1994). Cyn operon consists of three genes; cynT (cyanate permease), cynS (cyanase), and cynX (unknown hydrophobic protein) (Anderson et al. 1990). Guilloton et al. (1992) illustrated that cynT gene product was actually coded for carbonic anhydrase whose molecular weight was 24 kD. The enzyme was found to be in the oligomeric form in the solution and in the presence of bicarbonate, it partially dissociated from that structure. In cyn operon, the perceptible function of inducible carbonic anhydrase is to catalyze rapid hydration of carbon dioxide (formed due to decomposition of cyanate ion) to diffuse it out from the cell. In this way it helps the cell to replenish bicarbonate which is required by the cyanase enzyme to degrade cyanate ion. The second gene of cyn operon is the cynS gene which encodes for cyanase enzyme. CynS gene is usually activated only in the presence of high concentration of cyanate ion whereas arginine in excess acts as down regulator of this gene (Elleuche and Pöggeler 2008; Anderson et al. 1990).
The CynX is an unknown hydrophobic protein and not characterized till date. It is supposed to be a transporter protein for cyanate ion. Pao et al. (1998) included CynX as a member of major facilitator superfamily (MFS) of transporters. The size of the protein in E coli was found 393 amino acids size range which putatively might consist of 11 or 12 transmembrane structures. The protein has also exhibited considerable homology with the tartrate permease of Agrobacterium vitis and Bacillus subtilis at the sequence level (Pao et al. 1998). However, they were expressed at a much lower rate as compared to cynT and cynS gene.
At enzyme level, cyanase activity is regulated by the concentration of bicarbonate which acts as an inhibitor of cyanase enzyme. At low concentration, it shows uncompetitive inhibition. In that case, bicarbonate ions are bound to the anionic site of the active site and are interfered with cyanate ion binding. Whereas, at higher concentration of bicarbonate ions, the inactive complex formed at the active site and then degradation occurred to restart the binding action. This type of inactive rapid equilibrium formation between bicarbonate and cyanate ion is known as ping pong inhibition (Anderson and Little 1986).
Cyanide monooxygenase
Besides cyanases, two other enzymes are utilized by microbes for detoxification of cyanide i.e. cyanide monooxygenase and cyanide dioxygenase. Kunz et al. (1992) reported the presence of cyanide monooxygenase (also known as CNO) in Pseudomonas fluorescens NCIMB 11764 when the strain grew in cyanide solution (300 mM). In this case cyanide was degraded in two steps; first, it was converted to an intermediate formamide which then oxidatively cleaved to form formate and ammonia. The ammonia was then assimilated by conventional pathways (Kunz et al. 1992). The enzyme required pterin as cofactors (Fernandez et al. 2004). The formate produced in the aforesaid reaction was further oxidized to formic acid by formate dehydrogenase enzyme (FDH).
Cyanide dioxygenase
Cyanide dioxygenase enzyme converted cyanide into ammonia and carbon dioxide in a single step reaction. This enzyme also depends on pterin cofactor for its activity like cyanide monooxygenase. Cyanohydrins are also formed in cyanide dioxygenase mediated cyanide degradation pathways (Ebbs 2004).
Reductive pathway
The reductive pathway of cyanide degradation utilizes a well known enzyme called nitrogenase. This enzyme generally converts atmospheric nitrogen into ammonia and also be able to reduce cyanide into methane and ammonia. The first report of nitrogenase mediated reductive transformation of cyanide was reported by Hardy and Knight Jr (1967). An inducible nitrogenase from the strain Klebsiella oxytoca was isolated by Liu et al. (1997) after growing the strain in KCN. They transformed the strain with a nif containing plasmid and compared it with wild strain. The wild strain and transformed strain both exhibited diauxic growth in medium containing ammonium chloride and potassium cyanide. In culture broth, ammonium chloride was a preferred substrate over potassium cyanide and methane was detected as one of the main compounds in the growth medium (Liu et al. 1997). Another N2-fixing bacterium Azotobacter chroococcum NCIMB 8003 which contains nitrogenase enzyme was reported by Kelly (1968). He found that the strain was successful to reduce cyanide into ammonia by the action of the nitrogenase enzyme. Recently its genome was sequenced by Robson et al. (2015); however, no other cyanide metabolizing enzymes were found in this strain. In this context, a nitrogen fixing and strict aerobic strain of Azotobacter vinelandii was reported by Koksunan et al. 2013 and was extensively investigated for bioremediation of cyanide-contaminated wastewater. The strain A. vinelandii exhibited high nitrogenase expression as well as fast growth kinetics. The strain was first grown on nitrogen-free sucrose medium and its cyanide reduction ability was investigated. After that, the process was applied to remediate cyanide containing wastewater collected from cassava mill. A rapid cyanide reduction was observed in the early stationary growth phase rather than in the exponential phase and it was found that the cyanide concentration (4 mM-NaCN) was reduced by 69.7% within 66 h in 3L bioreactor (Koksunan et al. 2013).
Hydrolytic pathway
The hydrolytic pathway is the most widely exploited pathway for the degradation of cyanide by the microbes. Many hydrolases which are responsible for the degradation of inorganic as well as organic cyanides and nitriles have been studied extensively and reported in many literatures (Martínková et al. 2017; Dent et al. 2009; Jandhyala et al. 2005; Basile et al. 2008; Crum et al. 2016; O’Reilly and Turner 2003). Cyanidase, cyanide hydratase, formamidase, nitrilase, nitrile hydratase, amidase and thiocyanate hydrolase are the enzymes taking part in this pathway. In the hydrolytic pathway, direct cleavage of carbon nitrogen triple bond by the enzyme results in the formation of corresponding amide or acid and ammonia (Bhalla et al. 2012; Gupta et al. 2010).
Cyanidase and cyanide hydratase enzymes are highly specific in terms of their substrate specificity (cyanide). The former enzyme is mainly found in bacteria while later one is of fungal origin. Formamidase enzyme takes formamide as substrate and converts it into formic acid and ammonia. Nitrilase and amidase belong to Nitrilase superfamily of enzymes and convert organic nitriles and amide into their corresponding acids. Whereas, nitrile hydratase, a metalloenzyme help bacteria to use nitriles as nitrogen and carbon source. It catalyses the conversion of organic cyanide into an amide by adding water molecule to the cyano group which is further utilized as a substrate for amidase. Generally, in bacteria, the nitrile hydratase and amidase genes are cotranscribed and cotranslated. The enzyme thiocyanate hydrolase is responsible for catalyzing the reaction of thiocyanate to sulfate, ammonia and carbon dioxide (Gupta et al. 2010).
Cyanidase/cyanide dihydratase (EC 3.5.5.1)
Cyanidases (CDH) are cyanide degrading nitrilases which do not require any cofactor/co-substrate for their catalytic activity. Alcaligenes xylosoxidans subsp. denitrificans DF3 was reported to have this enzyme which hydrolyzed inorganic cyanide directly into formate without formation of any intermediate (Ingvorsen et al. 1991). Few other organisms, viz., Bacillus pumilus C1 (Meyers et al. 1991, 1993), Pseudomonas fluorescens NCIMB 11764 (Kunz et al. 1992) and P. stutzeri AK61 (Watanabe et al. 1998) were also reported which showed cyanidase activity when subjected to cyanide degradation. P. fluorescens NCIMB 11764 in this regard was quite unique because it has devised a different pathway for cyanide degradation. The strain possessed cyanide hydratase as well as cyanide dihydratase activities. The authors proposed that there may be a possibility that both the activities may be present in a single enzyme, but no detailed mechanism was proposed (Kunz et al. 1992). In another study, Martínková et al. (2015) investigated the bioremediation potentiality of Bacillus pumilus in the presence of cyanide waste. They found that cyanide degradation was improved when the process was carried out in alkaline pH. From that finding, they have engineered the strain by directed evolution combined with site-directed mutagenesis and got ample activity of these enzymes at pH 10. In an earlier report by Wang et al. (2012), demonstrated that mutants of Bacillus pumilus C1 cyanide dihydratase (CynDpum) showed improved activity at higher pH too (at pH 10).
The structural information of CDH enzyme was derived from P. stutzeri AK 61 homologue and it was reported that the enzyme was a homo-oligomeric spiral structured and comprising of 14 subunits with two fold symmetry (Sewell et al. 2003). In the case of Bacillus pumilus C1, it was found that cyanide dihydratase was consisting of 18 subunits and the fundamental structure was a dimer of molecular weight 37 kDa. It exhibited reversible, pH-dependent structural switching; at pH 8.0 the enzyme has a spiral structure and at pH 5.4 the enzyme subunit was reorganised and formed left handed helix. The putative amino acid sequence deduced from ORFs revealed that they shared high similarity with the cyanide dihydratase of Gram negative bacterium P. stutzeri AK61 (Jandhyala et al. 2003). The enzyme also exhibited similarity with nitrilases of fungal and bacterial origin (Thuku et al. 2007). It was also observed that C-terminal domain of cyanide dihydratase of Bacillus pumilus significantly influenced the thermostability and pH tolerance of the CDH enzyme because of its involvement in oligomerization at the interface of C surface (Crum et al. 2015).
An earlier report by Watanabe et al. (1998) described the cloning and expression of cyanidase gene from P. stutzeri AK61 to E. Coli. From the sequence data, the ORFs were searched and a protein consisting of 334 amino acids having a molecular weight of 37.5 kDa was predicted. Translated protein was shown to share 35.1% homology with nitrilase of Rhodococcus rhodochrous K22. Cyanide hydratase of Fusarium lateritium also exhibited homology up to 26.4%. The proposed active site had conserved cysteine residue like other nitrilase which played a pivotal role in catalysis. However the mechanism of action of cyanidase was not elucidated in detail; few other studies suggested that cysteine present in the active site might act as a nucleophile and could attack HCN, forming thioimidate as intermediate. Thioimidate was further metabolized to ammonia and an acyl product.
Cyanide hydratase (EC 4.2.1.66)
Cyanide hydratases belong to lyase superfamily and participated in cyanoamino acid metabolism. This enzyme was first reported in Stemphylium loti, a fungus which infected the common cyanogenic plant bird’s-foot trefoil (which is a low growing perennial legume) (Fry and Millar 1972). The cyanide hydratase mediated cyanide metabolism was restricted to fungi only and was not reported much in bacteria/plant/animals (Rinágelová et al. 2014). Most of the reported cyanide hydratases showed substrate specificity only for HCN, whereas nitrilases exhibited wide substrate specificity. Few fungi were also reported which could degrade metallocyanide; Fusarium solani and Fusarium oxysporum N-10 have been reported to degrade tetracyanonickelate (II) (Barclay et al. 1998; Yanase et al. 2000). Similarly, Fusarium lateritium also exhibited low cyanide hydratase activity with small aliphatic nitriles (acetonitrile and propionitrile) as well as with aromatic ones (benzonitrile) (Nolan et al. 2003). Other cyanide hydratase harboring microbes are Micromonospora braunnam (actinomycetes), Gloeocercospora sorghi, Leptosphaeria maculans, Aspergillus niger K10 (Wang and VanEtten 1992; Cluness et al. 1993). These cyanide hydratases have shown significant homology within these fungi at the amino acid level (Bhalla et al. 2012).
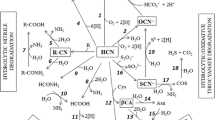
Recently we have proposed parts of the mechanism of cyanide hydratase enzyme from Serratia marcerans based on computational modeling and structural analysis (Kushwaha et al. 2018). The modeled structure exhibited a characteristic nitrilase superfamily catalytic triad consisting of E (at positions 45), K (at position 120) and C (at position 151). This modeled structure could reveal an active site within the range of 45 to176 amino acid residues containing catalytic triad cys-lys-glu (Fig. 1a). In the catalytic mechanism of cyanide degradation, cysteine was involved in the nucleophilic reaction occurred on the nitrile, resulting in the formation of a thioimidate. Lysine acted as a proton donor and caused hydrolysis; leading to the formation of tetrahedral intermediate. It results in C–S bond breakage and formamide was released. Glutamate acted as a proton acceptor which further caused secondary hydrolysis; leading to the formation of a second tetrahedral intermediate, which decomposed to form formic acid (Fig. 1b).

Mechanism of the cyanide hydratase activity. a Cyanide hydratase modelled structure of S. marcescens strain WW4 (Kushwaha et al. 2018). The catalytic site within the modelled structure consisted of a triad containing amino acid residues namely, a cysteine at position 151, lysine at position 120 and a glutamate at position 45. b Catalytic mechanism of cyanide hydratase: cysteine is involved in the nucleophilic attack on the nitrile which results in the formation of a thioimidate. Lysine acts as a proton donor. It causes breaking of the C–S bond leading to the release of formamide. Glutamate acts as a proton acceptor which then activates second hydrolysis leading to second tetrahedral intermediate, which decomposes to release formic acid (CHT cyanide hydratase, CynD cyanidedihydratase)
Substitution pathway
This pathway involves the transfer of a sulfur group from thiosulfate to cyanide and forming thiocyanate which is further assimilated subsequently as an alternate nitrogen source during microbial growth. The enzyme involved in such reactions belongs to sulfurtransferase family. The two important members of this family which take part in cyanide metabolism are rhodanese and mercaptopyruvate sulfurtransferase (Dash et al. 2009; Gupta et al. 2010; Bhalla et al. 2012).
Rhodanese (EC 2.8.1.1)
The rhodanese enzymes are ubiquitous enzymes and play a key role in sulfur metabolism. Rhodanese is also termed as thiosulfate sulfurtransferase. The enzyme transfers sulfur atom from donor to acceptor molecule. In cyanide metabolism, rhodanese enzymes help to transfer sulfur from thiosulfate (donor) to cyanide and less toxic thiocyanate molecule is formed (called ping pong mechanism) which is then assimilated in the cytoplasm of the organism. This enzyme has been reported in many eucaryotes and prokaryotes. Escherichia coli, Azotobacter vinelandii, Bacillus brevis, Fusarium sp., and many Thiobacillus sp. were reported to have rhodanese enzyme. Pseudomonas aeruginosa was reported to have constitutive rhodanese activity (Alexander and Volini 1987; Bordo and Bork 2002; Cipollone et al. 2004, 2006, 2008; Ezzi et al. 2003; Oyedeji et al. 2013; Wang and Volini 1968). Rhodanese enzymes are also used by many animals for cyanide detoxification in their metabolic pathway; e.g. Giant pandas absorbed more than 65% of cyanide by eating bamboo shoots (1 kg bamboo shoots contain 3.2 mg cyanide) and approximately 80% cyanide was converted to thiocyanate by rhodanese enzyme, which ultimately discharged through their urine (Huang et al. 2016).
Rhodanase activity and expression levels are dependent on the cyanide exposure to the tissue. Chaudhary and Gupta (2012) elucidated the catalytic activity of mitochondrial rhodanase of bovine. It was found that the enzyme consisted of 293 amino acids and possessed two equal sized domains, viz. N-terminal domain (inactive) and C-terminal domain (catalytic active). In the active site of rhodanase the sulfhydryl groups and aromatic groups were present in close proximity to each other. These aromatic groups have protected the catalytic sulfhydryl groups. The enzyme showed competitive inhibition in the presence of aromatic ions (aromatic sulfonates) but no inhibition was found in the presence of aliphatic ions which concluded that tryptophan was present in the active site. Gliubich et al. (1996) reported that the sulfhydryl group present in active site got oxidized and formed a sulfenyl group (–S–OH) when the rhodanase enzyme treated with hydrogen peroxide. This process has led to the inactivation of the enzyme. Other sulfhydryl group modifying agents like alkylating agents, aliphatic mercaptans, aromatic nitro compounds are also could eliminate the rhodanase activity. The alkylating agents are responsible for the substitution reaction with cysteinyl sulfur, while aromatic nitro compounds are responsible for intermolecular oxidation between two enzyme monomers. The aromatic mercaptans formed mixed disulfides with enzyme monomer (Wang and Volini 1968).
Mercaptopyruvate sulfurtransferase
Mercaptopyruvate sulfurtransferase enzymes belong to transferase enzyme family and are involved in many processes like iron sulfur clusters maintenance in protein (Alphey et al. 2003), molybdopterin cofactor biosynthesis for xanthine oxidase (Sörbo 1957), metabolism of selenium (Hannestad et al. 1981), thiamine and 4-thiouridine biosynthesis and cyanide detoxification (Ogata and Volini 1990). These enzymes are up-regulated when the organisms undergo stress condition, e.g. exposure to peroxide, hyposulphur conditions and infection by bacteriophage (Pagani et al. 1984).
These enzymes are widely distributed in a living system like rhodanase enzyme. They are also known as 3-mercaptopyruvate cyanide sulfurtransferase/β-mercaptopyruvate sulfurtransferase. This enzyme was first reported in trypanosomatid parasite Leishmania major (Williams et al. 2003) and found to be comprised of three domains; N-terminal domain, central domains and C-terminal domain. The active site of the enzyme contained conserved three residues Asp-61, His-75, and Ser-255 just like a serine protease triad and a cys residue in proximity at the Cys-253 position. The C terminal domain of enzyme was unique; consisting of 80-amino acids and involved in protein folding as well as in the interaction of sulfurtransferase with proteins (Nandi et al. 2000).
Conclusions
Cyanide containing toxins are usually generated from industrial plants. Many environmental and human health problems are caused by cyanide and its derivatives. Due to their high affinity towards metals, different metalloproteins get dysfunctional. Different strategies based on enzymatic cyanide degradation have been adopted and many useful enzymes (originated from different microbes and plants) such as cyanidase, cyanide hydratase, formamidase, nitrilase, nitrile hydratase, cyanide dioxygenase, cyanide monooxygenase, cyanase and nitrogenase and their mechanism of action have been studied extensively in this context. In order to understand the molecular mechanism of cyanide toxicity and to achieve efficient methods of their biodegradation, the next generation ‘omics’ technologies have been applied successfully during the last couple of decades. In the future, more advancement of omics tools and protein engineering based methods may help us to further strategize more efficient technologies to depollute the environment from the cyanide containing waste.
References
Acera F, Carmona MI, Castillo F, Quesada A, Blasco R (2017) A cyanide-induced 3-cyanoalanine nitrilase in the cyanide-assimilating bacterium Pseudomonas pseudoalcaligenes strain CECT 5344. Appl Environ Microbiol 83(9):e00089-17
Aichi M, Nishida I, Omata T (1998) Molecular cloning and characterization of a cDNA encoding cyanase from Arabidopsis thaliana. Plant Cell Physiol 39:S135
Akcil A (2003) Destruction of cyanide in gold mill effluents: biological versus chemical treatments. Biotechnol Adv 21:501–511
Akcil A, Mudder T (2003) Microbial destruction of cyanide wastes in gold mining: process review. Biotechnol Lett 25:445–450
Alexander K, Volini M (1987) Properties of an Escherichia coli rhodanese. J Biol Chem 262:6595–6604
Alphey MS, Williams RAM, Mottram JC et al (2003) The crystal structure of leishmania major 3-mercaptopyruvate sulfurtransferase A three-domain architecture with a serine protease-like triad at the active site. J Biol Chem 278:48219–48227
Anderson PM, Little RM (1986) Kinetic properties of cyanase. Biochemistry 25:1621–1626
Anderson PM, Sung Y, Fuchs JA (1990) The cyanase operon and cyanate metabolism. FEMS Microbiol Lett 87:247–252
Aronstein BN, Maka A, Srivastava VJ (1994) Chemical and biological removal of cyanides from aqueous and soil-containing systems. Appl Biochem Microbiol 41:700–707
ATSDR (2016) Releases report about exposure to PCBs at Oak Ridge Reservation, TN
Barac T, Taghavi S, Borremans B, Provoost A, Oeyen L, Colpaert JV, Vangronsveld J, van der Lelie D (2004) Engineered endophyticbacteria improve phytoremediation of water-soluble, volatile, organic pollutants. Nat Biotechnol 22:583–588
Barclay M, Hart A, Knowles CJ et al (1998) Biodegradation of metal cyanides by mixed and pure cultures of fungi. Enzyme Microb Technol 22:223–231
Basile LJ, Willson RC, Sewell BT, Benedik MJ (2008) Genome mining of cyanide-degrading nitrilases from filamentous fungi. Appl Microbiol Biotechnol 80(3):427–435
Baxter J, Cummings SP (2006) The current and future applications of microorganism in the bioremediation of cyanide contamination. Antonie Van Leeuwenhoek 90:1–17
Bhalla TC, Sharma N, Bhatia RK (2012) Microbial degradation of cyanides and nitriles. In: Satyanarayana T, Johri B, Prakash A (eds) Microorganisms in environmental management. Springer, Dordrecht, pp 569–587
Bhalla TC, Kumar V, Kumar V, Thakur N, Savitri (2018) Nitrile metabolizing enzymes in biocatalysis and biotransformation. Appl Biochem Biotechnol 185(4):925–946
Bordo D, Bork P (2002) The rhodanese/Cdc25 phosphatase superfamily: sequence–structure–function relations. EMBO Rep 3:741–746
Botz MM (2001) Overview of cyanide treatment methods, mining environmental management. Mining Journal Ltd., London, pp 28–30
Botz MM, Mudder TI, Akcil A (2015) Cyanide treatment: physical, chemical and biological processes. In: Adams M (ed) Advances in gold ore processing. Elsevier, Amsterdam, pp 672–700
Cabello P, Luque-Almagro VM, Olaya-Abril A et al (2018) Assimilation of cyanide and cyano-derivatives by Pseudomonas pseudoalcaligenes CECT5344: from omic approaches to biotechnological applications. FEMS Microbiol Lett 365(6):fny032
Campos MG, Pereira P, Roseiro JC (2006) Packed-bed reactor for the integrated biodegradation of cyanide and formamide by immobilised Fusarium oxysporum CCMI 876 and Methylobacterium sp. RXM CCMI 908. Enzyme Microb Technol 38:848–854
Castrıc PA, Farnden KJF, Conn EE (1972) Cyanide metabolism in higher plants. V. The formation of asparagine from Ii-cyanoalanine. Arch Biochem Biophys 152:62–69
Chaudhary M, Gupta R (2012) Cyanide detoxifying enzyme: rhodanese. Curr Biotechnol 1:327–335
Cipollone R, Bigotti MG, Frangipani E et al (2004) Characterization of a rhodanese from the cyanogenic bacterium Pseudomonas aeruginosa. Biochem Biophys Res Commun 325:85–90
Cipollone R, Ascenzi P, Frangipani E, Visca P (2006) Cyanide detoxification by recombinant bacterial rhodanese. Chemosphere 63:942–949
Cipollone R, Ascenzi P, Tomao P, Imperi F, Visca P (2008) Enzymatic detoxification of cyanide: clues from Pseudomonas aeruginosa rhodanese. J Mol Microbiol Biotechnol 15:199–211
Cluness MJ, Turner PD, Clements E et al (1993) Purification and properties of cyanide hydratase from Fusarium lateritium and analysis of the corresponding chy1 gene. Microbiology 139:1807–1815
Cobbett CS, Goldsbrough PB (2000) Mechanisms of metal resistance: phytochelatins and metallothioneins. In: Raskin I, Ensley BD (eds) Phytoremediation of toxic metals using plants to clean up the environment. Wiley, New York, pp 247–271
Crum MA, Park JM, Sewell BT, Benedik MJ (2015) C-terminal hybrid mutant of Bacillus pumilus cyanide dihydratase dramatically enhances thermal stability and pH tolerance by reinforcing oligomerization. J Appl Microbiol 118:881–889
Crum MA, Sewell BT, Benedik MJ (2016) Bacillus pumilus cyanide dihydratase mutants with higher catalytic activity. Front Microbiol. 7:1264
Dash RR, Gaur A, Balomajumder C (2009) Cyanide in industrial wastewaters and its removal: a review on biotreatment. J Hazard Mater 163:1–11
Dent KC, Weber BW, Benedik MJ, Sewell BT (2009) The cyanide hydratase from Neurospora crassa forms a helix which has a dimeric repeat. Appl Microbiol Biotechnol 82:271–278
Ebbs S (2004) Biological degradation of cyanide compounds. Curr Opin Biotechnol 15:231–236
Ebel M, Evangelou MWH, Schaeffer A (2007) Cyanide phytoremediation by water hyacinths (Eichhornia crassipes). Chemosphere 66:816–823
Elleuche S, Pöggeler S (2008) A cyanase is transcriptionally regulated by arginine and involved in cyanate decomposition in Sordaria macrospora. Fungal Genet Biol 45:1458–1469
Emmanuel OA, Emmanuel NU (1981) Characterization of rhodanese from cassava leaves and tubers. J Exp Bot 32(5):1021–1027
Ezzi MI, Pascual JA, Gould BJ, Lynch JM (2003) Characterisation of the rhodanese enzyme in Trichoderma spp. Enzyme Microb Technol 32:629–634
Fallon RD (1992) Evidence of a hydrolytic route for anaerobic cyanide degradation. Appl Environ Microbiol 58(9):3163–3164
Fernandez RF, Dolghih E, Kunz DA (2004) Enzymatic assimilation of cyanide via pterin-dependent oxygenolytic cleavage to ammonia and formate in Pseudomonas fluorescens NCIMB 11764. Appl Environ Microbiol 70:121–128
Frapolli M, Pothier JF, Défago G, Moënne-Loccoz Y (2012) Evolutionary history of synthesis pathway genes for phloroglucinol and cyanide antimicrobials in plant-associated fluorescent pseudomonads. Mol Phylogenet Evol 63:877–890
Fry WE, Millar RL (1972) Cyanide degradion by an enzyme from Stemphylium loti. Arch Biochem Biophys 151:468–474
Gliubich F, Gazerro M, Zanotti G et al (1996) Active site structural features for chemically modified forms of rhodanese. J Biol Chem 271:21054–21061
Guilloton MB, Korte JJ, Lamblin AF et al (1992) Carbonic anhydrase in Escherichia coli. A product of the cyn operon. J Biol Chem 267:3731–3734
Gupta N, Balomajumder C, Agarwal VK (2010) Enzymatic mechanism and biochemistry for cyanide degradation: a review. J Hazard Mater 176:1–13
Hannestad U, Mårtensson J, Sjödahl R, Sörbo B (1981) 3-Mercaptolactate cysteine disulfiduria: biochemical studies on affected and unaffected members of a family. Biochem Med 26:106–114
Harano Y, Suzuki I, Maeda S et al (1997) Identification and nitrogen regulation of the cyanase gene from the cyanobacteria Synechocystis sp. strain PCC 6803 and Synechococcus sp. strain PCC 7942. J Bacteriol 179:5744–5750
Hardy RWF, Knight E Jr (1967) ATP-dependent reduction of azide and HCN by N2-fixing enzymes of Azotobacter vinelandii and Clostridium pasteurianum. Biochim Biophys Acta (BBA)-Enzymol 139:69–90
He J, Kappler A (2017) Recovery of precious metals from waste streams. Microb Biotechnol 10(5):1194–1198
Huang H, Yie S, Liu Y et al (2016) Dietary resources shape the adaptive changes of cyanide detoxification function in giant panda (Ailuropoda melanoleuca). Sci Rep 6:34700
Huertas MJ, Luque-Almagro VM, Martínez-Luque M et al (2006) Cyanide metabolism of Pseudomonas pseudoalcaligenes CECT5344: role of siderophores. Biochem Soc Trans 34(1):152–155
Ibáñez MI, Cabello P, Luque-Almagro VM, Sáez LP, Olaya A, Sánchez de Medina V et al (2017) Quantitative proteomic analysis of Pseudomonas pseudoalcaligenes CECT5344 in response to industrial cyanide-containing wastewaters using Liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS). PLoS ONE 12(3):e0172908
Ingvorsen K, Højer-Pedersen B, Godtfredsen SE (1991) Novel cyanide-hydrolyzing enzyme from Alcaligenes xylosoxidans subsp. denitrificans. Appl Environ Microbiol 57:1783–1789
Jandhyala DM, Berman M, Meyers PR, Sewell BT, Willson RC, Benedik MJ (2003) CynD, the cyanide dihydratase from Bacillus pumilus: gene cloning and structural studies. Appl Environ Microbiol 69:4794–4805
Jandhyala DM, Willson RC, Sewell BT, Benedik MJ (2005) Comparison of cyanide-degrading nitrilases. Appl Microbiol Biotechnol 68(3):327–335
Kaewkannetra P, Imai T, Garcia-Garcia FJ, Chiu TY (2009) Cyanide removal from cassava mill wastewater using Azotobactor vinelandii TISTR 1094 with mixed microorganisms in activated sludge treatment system. J Hazard Mater 172:224–228
Kang DH, Hong LY, Paul Schwab A, Banks MK (2007) Removal of Prussian blue from contaminated soil in the rhizosphere of cyanogenic plants. Chemosphere 69:1492–1498
Kebeish R, Al-Zoubi O (2017) Expression of the cyanobacterial enzyme cyanase increases cyanate metabolism and cyanate tolerance in Arabidopsis. Environ Sci Pollut Res Int 24(12):11825–11835
Kelly M (1968) The kinetics of the reduction of isocyanides, acetylenes and the cyanide ion by nitrogenase preparation from Azotobacter chroococcum and the effects of inhibitors. Biochem J 107:1–6
Koeth RA, Kalantar-Zadeh K, Wang Z et al (2013) Protein carbamylation predicts mortality in ESRD. J Am Soc Nephrol 24:853–861
Koksunan S, Vichitphan S, Laopaiboon L et al (2013) Growth and cyanide degradation of Azotobacter vinelandii in cyanide-containing wastewater system. J Microbiol Biotechnol 23:572–578
Kumar V, Kumar V, Bhalla TC (2013) In vitro cyanide degradation by Serretia marcescens RL2b. Int J Environ Sci 3:1969
Kumar R, Saha S, Dhaka S et al (2017) Remediation of cyanide-contaminated environments through microbes and plants: a review of current knowledge and future perspectives. Geosystem Eng 20:28–40
Kunz DA, Nagappan O, Silva-Avalos J, Delong GT (1992) Utilization of cyanide as nitrogenous substrate by Pseudomonas fluorescens NCIMB 11764: evidence for multiple pathways of metabolic conversion. Appl Environ Microbiol 58:2022–2029
Kushwaha M, Kumar V, Mahajan R et al (2018) Molecular insights into the activity and mechanism of cyanide hydratase enzyme associated with cyanide biodegradation by Serratia marcescens. Arch Microbiol. https://doi.org/10.1007/s00203-018-1524-0
Lamblin AF, Fuchs JA (1994) Functional analysis of the Escherichia coli K-12 cyn operon transcriptional regulation. J Bacteriol 176:6613–6622
Larsen M, Trapp S, Pirandello A (2004) Removal of cyanide by woody plants. Chemosphere 54:325–333
Liu JK, Liu CH, Lin CS (1997) The role of nitrogenase in a cyanide-degrading Klebsiella oxytoca strain. Proc Natl Sci Counc Repub China B 21:37–42
Lovecka P, Thimova M, Grznarova P et al (2015) Study of Cytotoxic Effects of Benzonitrile pesticides. Biomed Res Int 2015:381264
Luque-Almagro VM, Huertas M-J, Sáez LP et al (2008) Characterization of the Pseudomonas pseudoalcaligenes CECT5344 cyanase, an enzyme that is not essential for cyanide assimilation. Appl Environ Microbiol 74:6280–6288
Luque-Almagro VM, Blasco R, Martínez-Luque M et al (2011) Bacterial cyanide degradation is under review: Pseudomonas pseudoalcaligenes CECT5344, a case of an alkaliphilic cyanotroph. Biochem Soc Trans 39(1):269–274
Luque-Almagro VM, Moreno-Vivián C, Roldán MD (2016) Biodegradation of cyanide wastes from mining and jewellery industries. Curr Opin Biotechnol 38:9–13
Luque-Almagro VM, Cabello P, Sáez LP, Olaya-Abril A, Moreno-Vivián C, Olden MD (2018) Exploring anaerobic environments for cyanide and cyano-derivatives microbial degradation. Appl Microbiol Biotechnol 102(3):1067–1074
Maniyam MN, Sjahrir F, Ibrahim AL, Cass AEG (2013) Biodegradation of cyanide by Rhodococcus UKMP-5M. Biologia 68(2):177–185
Manso Cobos I, Ibáñez García MI, de la Peña Moreno F et al (2015) Pseudomonas pseudoalcaligenes CECT5344, a cyanide-degrading bacterium with by-product (polyhydroxyalkanoates) formation capacity. Microb Cell Fact 14:77
Martínková L, Veselá AB, Rinágelová A, Chmátal M (2015) Cyanide hydratases and cyanide dihydratases: emerging tools in the biodegradation and biodetection of cyanide. Appl Microbiol Biotechnol 99(21):8875–8882
Martínková L, Rucká L, Nešvera J, Pátek M (2017) Recent advances and challenges in the heterologous production of microbial nitrilases for biocatalytic applications. World J Microbiol Biotechnol 33(1):8
Maruyama A, Saito K, Ishizawa K (2001) β-cyanoalanine synthase and cysteine synthase from potato: molecular cloning, biochemical characterization, and spatial and hormonal regulation. Plant Mol Biol 46:749–760
Mccutcheon SC, Schnoor JL (2003) Phytoremediation: transformation and control of contaminants. Wiley, New York, pp 663–694
Mekuto L, Alegbeleye OO, Ntwampe SKO et al (2016) Co-metabolism of thiocyanate and free cyanide by Exiguobacterium acetylicum and Bacillus marisflavi under alkaline conditions. 3 Biotech 6:173
Meyers PR, Gokool P, Rawlings DE, Woods DR (1991) An efficient cyanide-degrading Bacillus pumilus strain. Microbiology 137:1397–1400
Meyers PR, Rawlings DE, Woods DR, Lindsey GG (1993) Isolation and characterization of a cyanide dihydratase for Bacillus pumilus C1. J Bacteriol 175:6105–6112
Miller JM, Conn EE (1980) Metabolism of hydrogen cyanide by higher plants. Plant Physiol 65:1199–1202
Møller BL (2010) Functional diversifications of cyanogenic glucosides. Curr Opin Plant Biol 13:337–346
Myers DF, Fry WE (1978) Enzymatic release and metabolism of hydrogen cyanide in sorghum infected with Gloeocercospora sorghi. Phytopathology 68:1717–1722
Nandi DL, Horowitz PM, Westley J (2000) Rhodanese as a thioredoxin oxidase. Int J Biochem Cell Biol 32:465–473
Nolan LM, Harnedy PA, Turner P et al (2003) The cyanide hydratase enzyme of Fusarium lateritium also has nitrilase activity. FEMS Microbiol Lett 221:161–165
Ogata K, Volini M (1990) Mitochondrial rhodanese: membrane-bound and complexed activity. J Biol Chem 265:8087–8093
O’Reilly C, Turner PD (2003) The nitrilase family of CN hydrolysing enzymes—a comparative study. J Appl Microbiol 95(6):1161–1174
Oyedeji O, Awojobi KO, Okonji RE, Olusola OO (2013) Characterization of rhodanese produced by Pseudomonas aeruginosa and Bacillus brevis isolated from soil of cassava processing site. Afr J Biotechnol 12(10):1104–1114
Pagani S, Bonomi F, Cerletti P (1984) Enzymic synthesis of the iron-sulfur cluster of spinach ferredoxin. Eur J Biochem 142:361–366
Panter KE (2018) Cyanogenic glycoside–containing plants. In: Gupta RC (ed) Veterinary toxicology, 3rd edn. Elsevier, Amsterdam, pp 935–940
Pao SS, Paulsen IT, Saier MH (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Park JM, Sewell BT, Benedik MJ (2017) Cyanide bioremediation: the potential of engineered nitrilases. Appl Microbiol Biotechnol 101:3029–3042
Paschka MG, Ghosh RS, Dzombak DA (1999) Potential water-quality effects from iron cyanide anticaking agents in road salt. Water Environ Res 71:1235–1239
Pickering IJ, Prince RC, George MJ, Smith RD, George GN, Salt DE (2000) Reduction and coordination of arsenic in Indian mustard. Plant Physiol 122:1171–1177
Piotrowski M, Schonfelder S, Weiler EW (2001) The Arabidopsis thaliana isogene NIT4 and its orthologs in tobacco encode beta-cyano-l-alanine hydratase/nitrilase. J Biol Chem 276:2616–2621
Rinágelová A, Kaplan O, Veselá AB et al (2014) Cyanide hydratase from Aspergillus niger K10: overproduction in Escherichia coli, purification, characterization and use in continuous cyanide degradation. Process Biochem 49:445–450
Robson RL, Jones R, Robson RM et al (2015) Azotobacter genomes: the genome of Azotobacter chroococcum NCIMB 8003 (ATCC 4412). PLoS ONE 10:e0127997
Samiotakis M, Ebbs SD (2004) Possible evidence for transport of an iron cyanide complex by plants. Environ Pollut 127:169–173
Sewell BT, Berman MN, Meyers PR et al (2003) The cyanide degrading nitrilase from Pseudomonas stutzeri AK61 is a two-fold symmetric, 14-subunit spiral. Structure 11:1413–1422
Sharma M, Sharma NN, Bhalla TC (2005) Hydroxynitrile lyases: at the interface of biology and chemistry. Enzyme Microb Technol 37:279–294
Shirai R (1978) Study on cyanide metabolizing activity in mesocarp of Rosaceae. J Coll Arts Sci Chiba Univ B-11:11–33
Siciliano SD, Germida JJ (1998) Mechanisms of phytoremediation: biochemical and ecological interactions between plants and bacteria. Environ Rev 6:65–79
Singh U, Arora NK, Sachan P (2018) Simultaneous biodegradation of phenol and cyanide present in coke-oven effluent using immobilized Pseudomonas putida and Pseudomonas stutzeri. Braz J Microbiol 49(1):38–44
Sörbo B (1957) Enzymic transfer of sulfur from mercaptopyruvate to sulfite or sulfinates. Biochim Biophys Acta 24:324–329
Sung Y-C, Fuchs JA (1992) The Escherichia coli K-12 cyn operon is positively regulated by a member of the lysR family. J Bacteriol 174:3645–3650
Taiz L, Zeiger E (2002) Plant physiology. Sunderland, Sinauer, p 690
Thuku RN, Weber BW, Varsani A, Sewell BT (2007) Post-translational cleavage of recombinantly expressed nitrilase from Rhodococcus rhodochrous J1 yields a stable, active helical form. FEBS J 274:2099–2108
Tomati U, Federıcı G, Cannella C (1972) Rhodanese activity in chloroplasts. Physiol Chem Physics 4:193–196
Trapp S, McFarlane C (eds) (1995) Plant contamination: modeling and simulation of organic processes. Lewis, Boca Raton, p 254
Volkering F, Breure AM, Rulkens WH (1998) Microbiological aspects of surfactant use for biological soil remediation. Biodegradation 8:401–417
Wang P, VanEtten HD (1992) Cloning and properties of a cyanide hydratase gene from the phytopathogenic fungus Gloeocercospora sorghi. Biochem Biophys Res Commun 187:1048–1054
Wang SF, Volini M (1968) Studies on the active site of rhodanese. J Biol Chem 243:5465–5470
Wang L, Watermeyer JM, Mulelu AE, Sewell BT, Benedik MJ (2012) Engineering pH-tolerant mutants of a cyanide dihydratase. Appl Microbiol Biotechnol 94(1):131–140
Watanabe A, Yano K, Ikebukuro K, Karube I (1998) Cyanide hydrolysis in a cyanide-degrading bacterium, Pseudomonas stutzeri AK61, by cyanidase. Microbiology 144:1677–1682
White DM, Schnabel W (1998) Treatment of cyanide waste in a sequencing batch biofilm reactor. Water Res 32:254–257
Williams RAM, Kelly SM, Mottram JC, Coombs GH (2003) 3-Mercaptopyruvate sulfurtransferase of leishmania contains an unusual C-terminal extension and is involved in thioredoxin and antioxidant metabolism. J Biol Chem 278:1480–1486
Yanase H, Sakamoto A, Okamoto K et al (2000) Degradation of the metal-cyano complex tetracyanonickelate (II) by Fusarium oxysporum N-10. Appl Microbiol Biotechnol 53:328–334
Young RA (1954) Flavor qualities of some edible oriental bamboos. Econ Bot 8:377–386
Yu XZ, Gu JD (2007) Differences in Michaelis-Menten kinetics for different cultivars of maize during cyanide removal. Exotoxicol Environ Saf 67:254–259
Yu XZ, Gu JD (2009) Uptake, accumulation and metabolic response of ferricyanide in weeping willows. J Environ Monit 11(1):145–152
Yu X, Trapp S, Zhou P et al (2004) Metabolism of cyanide by Chinese vegetation. Chemosphere 56:121–126
Yu X, Trapp S, Zhou P (2005a) Phytotoxicity of cyanide to weeping willow trees. Environ Sci Pollut Res 12:109–113
Yu X, Trapp S, Zhou P, Hu H (2005b) The effect of temperature on the rate of cyanide metabolism of two woody plants. Chemosphere 59:1099–1104
Zagrobelny M, Bak S, Olsen CE, Møller BL (2007) Intimate roles for cyanogenic glucosides in the life cycle of Zygaena filipendulae (Lepidoptera, Zygaenidae). Insect Biochem Mol Biol 37:1189–1197
Zagrobelny M, Scheibye-Alsing K, Jensen NB et al (2009) 454 pyrosequencing based transcriptome analysis of Zygaena filipendulae with focus on genes involved in biosynthesis of cyanogenic glucosides. BMC Genom 10:574
Acknowledgements
We are thankful to Prof Ian S. Maddox, who has kindly invited us to contribute this article and also for his encouragement and advice since the invitation. We are also thankful to reviewers and editors for their valuable suggestions. Research in MS lab is supported by Uttar Pradesh Council of Science and Technology (Govt. of Uttar Pradesh province, India). Research in YA lab is supported by Indian Council of Medical Research and Department of Biotechnology (Ministry of Science & Technology, Govt. of India). SC gratefully acknowledges the Council of Scientific and Industrial Research (CSIR), India for an extramural research grant. Authors would like to thank the technical English translation expert Dr K B S Krishna, Assistant Professor, Department of English and European Languages, Central University of Himachal Pradesh, Dharamshala for his inputs and proofreading.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharma, M., Akhter, Y. & Chatterjee, S. A review on remediation of cyanide containing industrial wastes using biological systems with special reference to enzymatic degradation. World J Microbiol Biotechnol 35, 70 (2019). https://doi.org/10.1007/s11274-019-2643-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2643-8