
Abstract
Tilletia controversa Kühn (TCK) is an important quarantine pathogen that causes wheat dwarf bunt and results in devastating damage to wheat production. The fungus is difficult to be distinguished from T. caries and T. laevis, which cause wheat common bunt, based on morphological, physiological and symptomatological characteristics of the pathogens. The traditional detection of the fungus can be a long and tedious process with poor accuracy. The inter-simple sequence repeat (ISSR) technique has been used for identifying molecular markers for detection of TCK. Of 28 ISSR primers screened, ISSR-859 amplified a specific 678 bp DNA fragment from all TCK isolates but not from any isolates of the common bunt fungi or other pathogenic fungi tested. Based on the fragment sequence, a pair of sequence characterized amplified region (SCAR) primers was designed, which amplified a 372 bp DNA fragment specifically in TCK. The SCAR marker was detected using as low as 1 ng template DNA of TCK, and was also detected using broken teliospores and DNA from asymptomatic wheat samples. We developed the SYBR Green I and TaqMan Green I and TaqMan real-time polymorphism chain reaction methods to detect TCK with the detection limit of 0.1 fg with asymptomatic wheat samples. Further work is needed to develop a rapid test kit for this pathogenic fungus using the designed specific primers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat dwarf bunt is a destructive disease on wheat. Its causal fungus, Tilletia controversa J. G. Kühn (TCK) (Hoffmann 1982; Trione 1982), is an internationally important quarantine pathogen. A number of countries have placed restrictions on the entry of TCK-contaminated wheat grain. By 1998, 15 countries had documented regulations against the entry of TCK (United States Department of Agriculture 1998). These countries primarily concern that importing wheat grain containing TCK could lead to the introduction and establishment of the disease. In international trades, TCK may spread from one country to another mainly through teliospores contaminated in grain. As a potential invading species, if TCK spreads to a new environment where protective measures are generally lacking, TCK would cause serious problems in wheat production and could be difficult to control. Therefore, the most important step is to establish an effective detection method to prevent the pathogen from entering into countries like China without the pathogen.
Its closely related species, T. caries (DC.) Tul. & C. Tul. [syn. T. tritici (Bjerk.) G. Winter] and T. laevis J. G. Kühn (syn. T. foetida (Wallr.) Liro] causing wheat common bunt, are more widely distributed in the world. The three Tilletia species are morphologically so similar that when TCK was first discovered in Montana, the US, it was considered to be a variant of T. caries. The pathogen was later identified as a new species (Young 1935). The traditional methods for diagnosis and detection of TCK and related species were mostly based on microscopic examination and germination of teliospores (Baylis 1958; Meiners and Waldher 1959; Duran and Fishcher 1961; Hess and Frione 1986), which were not suitable for rapid detection of a large number of samples, as it takes almost 20 days to germinate teliospores of the species. The morphological diagnosis may not produce accurate results. For the purpose of quarantine, developing a rapid and accurate method for detecting TCK is a high priority.
With the continuous development of biotechnology, molecular detection has become an effective approach for distinguishing among different species. Compared with the traditional diagnostic methods, molecular detection has several potential advantages, as it is faster and more reliable. In the recent years, studies on Tilletia species mainly focused on analyzing the relationships between TCK and its closely related species using molecular markers, such as restriction fragment length polymorphism (RFLP) (Pimentel et al. 1998), random amplified polymorphism DNA (RAPD) (Pimentel 2000). However, the techniques used in these studies could not distinguish among TCK, T. caries and T. laevis. Kochanova et al. (2004) developed certain polymorphism chain reaction (PCR) primers for detecting TCK and T. caries. Liu et al. (2009) used SCAR markers by AFLP to differentiate the species. Yuan et al. (2009) used RAPD primer-mediated asymmetric-PCR (RM-PCR) to differentiate TCK from T. caries. McDonald et al. (2000) and Zupunski et al. (2011) used the repetitive-sequence-based polymerase chain reaction (rep-PCR) fingerprinting technique to determine the diversity of pathogens. Although some of the studies showed that molecular diagnosis is promising, a robust and much more sensitive detection method is still needed for quick detection of TCK.
Inter-simple sequence repeat (ISSR) fingerprinting is a robust, PCR-based technique for DNA amplification of putative microsatellite regions (Zietkiewicz et al. 1994). Microsatellites or simple sequence repeats (SSRs) are repeated arrays of short motifs of 1–4 base pairs in length. These DNA regions are characterized by their hypervariability, reproducibility, Mendelian inheritance, codominant nature and abundance in the genomes of nearly all eukaryotes (Scott et al. 2000). The ISSR technique can be undertaken for any species that contains a sufficient number and distribution of SSR motifs and has the advantage that genomic sequence data is not required (Gupta et al. 1994). Therefore, it has been widely employed as DNA markers for genetic polymorphism analysis in the field of population genetics (Lian et al. 2001; Menzies et al. 2003; Talhinhas et al. 2003), genetic map construction (Kojima et al. 1998; Davila et al. 1999; Arcade et al. 2000) and detection of biological invasion (Meekins et al. 2001; Sun et al. 2005). Compared with other fingerprinting methods such as RAPD, RFLP and AFLP, the ISSR technique was an effective method by its simplicity, rapidness, cheap cost and high polymorphisms characteristic and was suitable for use in molecular detection. In this study, we applied the ISSR technique in identification of molecular markers for distinguishing TCK and related fungal species and converted a TCK-specific ISSP marker to a sequence characterized amplified region (SCAR) marker and also developed SYBR Green I and TaqMan real-time PCR for detection and diagnosis of TCK.
Materials and methods
Fungal isolates and DNA extraction
A list of isolates of TCK, related bunt and smut fungal species (T. caries, T. laevis, Ustilago tritici, U. hordei, U. scitaminea, U. maydis, Neovossia horrida, Sphacelotheca sorghi and S. reiliana) and some other common fungal pathogens of wheat used in this study is given in Table 1. The isolates were obtained from the United States, China and the Czech Republic. Genomic DNA was extracted from 10 mg of teliospores using a protocol reported by Liu et al. (2009). DNA was quantified using agarose gel electrophoresis and stored at −20 °C.
ISSR-PCR amplification
To identify differential fragments, the genomic DNA of the TCK isolates and its related species were amplified using 40 ISSR primers (Table 2), which were designed by the University of British Columbia (http://www.michaelsmith.ubc.ca/services/NAPS/Primer_Sets). The primers were synthesized at Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China). Amplifications were performed in a total volume of 25 μL reaction containing 1× PCR buffer (Tiangen, Beijing, China), 2 mM of MaCl2 (Tiangen, Beijing, China), 1.2 μM of primer (Tiangen, Beijing, China), 0.1 mM of dNTPs (Tiangen, Beijing, China), 1 unit of Taq polymerase (Tiangen, Beijing, China) and 30 ng of genomic DNA. A PTC-220 thermal cycler (MJ Research, Inc., St. Bruno, Quebec, Canada) was used with the following cycling conditions: a denaturing cycle of 94 °C for 1 min; 40 cycles of 94 °C for 1 min, 42–55 °C depending upon primers (Table 2) for 1 min, and 72 °C for 2 min; and a 3 min extension at 72 °C. Amplification products were detected by electrophoresis on 1.5 % (w/v) agarose gels stained in ethidium bromide and visualized using a u.v. transilluminator (Bio-Rad Gel Doc XR, Hercules, CA, USA).
DNA sequencing and development of SCAR markers
A band specific for TCK amplified with primer UBC_859 was purified using an AxyPrep DNA Gel Extraction Kit (Axygen Scientific, Union City, CA, USA) according to the manufacturer’s instructions. The purified DNA product was ligated into a pMD 18-T vector (TaKaRa Biotech, Dalian, China), and the cloned fragment was sequenced by Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China). The sequence was blasted against GenBank databases to check for potential homologies. Based on the sequence, a pair of SCAR primers (ISSR140A/ISSR511A) was designed using the DNAMAN software.
Tests for SCAR marker specificity
The specificity of the SCAR marker was tested with genomic DNA of the fungal isolates in Table 1. Amplifications were performed in a total volume 25 μL reaction containing 1× PCR buffer (Tiangen, Beijing, China), 2 mM of MaCl2 (Tiangen, Beijing, China), 0.4 μM of each primer (Tiangen, Beijing, China), 0.12 mM of dNTPs (Tiangen, Beijing, China), 0.75 unit of Taq polymerase (Tiangen, Beijing, China) and 10 ng of genomic DNA. A PTC-220 thermal cycler (MJ Research, Inc.) was used with the following cycling conditions: a denaturing cycle of 94 °C for 5 min; 30 cycles of 94 °C for 30 s, 58 °C 30 s and 72 °C for 1 min; and a 10 min extension at 72 °C. We checked the DNA of T. controversa, T. caries and T. laevis by ITS primers (ITS4: 5′TCCTCCGCTTATTGATATGC3′; ITS5: 5′GGAAGTAAAAGTCGTAA CAAGG3′) and the specificity of the SCAR marker by a multiplex PCR test.
Tests for SCAR marker sensitivity
The sensitivity of the SCAR marker was tested with purified genomic DNA of TCK, which was serially diluted as the following concentrations: 100 ng/μL, 50 ng/μL, 20 ng/μL, 10 ng/μL, 5 ng/μL, 1 ng/μL, 100 pg/μL, 10 pg/μL, 1 pg/μL and 0.1 pg/μL. One microliter of each dilution was used in PCR. The reaction mix and amplification conditions were the same as mentioned above.
In addition, we used 1, 2, 3, 5, 10, 12 and 20 TCK teliospores which were broken using needles. The broken teliospores were directly used as templates in PCR amplification with the SCAR primers to check the sensitivity of detecting TCK.
Detection of wheat samples using the SCAR marker
The asymptomatic wheat samples were collected from inoculated plants grown in a controlled greenhouse and tested with the SCAR marker. The samples included different growth stages of wheat (roots and 1-leaf, 2-leaf, 3-leaf, tillering, jointing and mature stages). DNA was extracted from the leaf samples. The DNA of non-inoculated plants was used as a negative control. Five biological repeats were used for each stage sample as well as the healthy wheat control.
RT-PCR detection
The fluorescent-based SYBR Green and TaqMan were used in the real-time PCR (RT-PCR) experiment. The primer pair (5′ ACGACCGACTTTCCGAGAGC 3′/5′ GTGTGGGACGAAGGCATCAA 3′) and TaqMan probe (FAM 5′-ACGACTTGCGGTCCCTCCACA-3′TAMAR) were designed using the Primer 5.0 software and synthesized by Shanghai Shenggong Biological Engineering Technology and Service Co., Ltd., China.
For SYBR Green, the primer pair was used and the PCR reaction was performed in 20 μL reaction mixture containing 1× PCR buffer (CWbio, China), 0.4 μM/L of primers and 2 μL of plasmid DNA sample. The ABI7500 was used with the following programme: 95 °C for 10 min, 40 cycles each at 95 °C for 15 s and 60 °C for 60 s. The fluorescent signal was collected at the annealing step and the extending step (60 °C for 60 s) of each cycle.
The TaqMan PCR reaction was optimized in 20 μL reaction mixture containing 2× GoldStar TaqMan Mixture (with ROX) (CWbio, China), 0.4 μM/L of each primer, 0.2 μM/L TaqMan probe and 2 μL of plasmid DNA sample. The ABI7500 (Applied Biosystems, USA) was used with the following programme for DNA amplification: 95 °C for 10 min, 40 cycles each at 95 °C for 15 s and 60 °C for 60 s. A standard curve for RT-PCR was established according to Yuan et al. (2009) except with the pUCm-T vector instead of pMD18-T. The DNA of teliospores of 13 TCK isolates and 9 T. caries isolates was amplified using TaqMan RT-PCR. DNA of non-inoculated wheat was used as a negative control.
In order to investigate the dynamic changes of the titer of TCK in host plants throughout the year, the artificially contaminated wheat seeds with TCK were sown in experimental fields. The asymptomatic wheat plants at different growth stages were tested using TaqMan RT-PCR.
Results
Identification of a TCK-specific ISSR marker
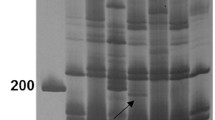
From the 40 ISSR primers screened, primer UBC_859 produced a fragment of about 680 bp in the TCK isolates, but not in those of T. caries and T. laevis (Fig. 1). Thus, the amplified fragment was specific to TCK.

Selection of a specific fragment of T. controversa J. G. Kühn (TCK) with an inter-simple sequence repeat (ISSR) primer UBC_859. Lanes 1–8, isolates TCK1–TCK8; lanes 9–12 T. laevis isolates TF1–TF4; lanes 13–16 T. caries isolates TCT1–TCT4; and M marker DL2000 marked with fragment sizes in base pair (bp). The specific band is marked with an arrow
Development of a SCAR marker
The sequencing result showed that the length of the ISSR marker was 678 bp (Fig. 2). No sequences in GenBank were found to have a significant homology with the ISSR marker sequence. Based on the sequence, a pair of primers, ISSR140A: 5′-TGGTGGTCGGGAAAGATTAGA-3′ and ISSR511A: 5′-GGGACGAAGGCATCAAGAAG-3′ was designed to develop a SCAR marker for TCK. The pair of primers amplified a 372 bp fragment with TCK isolates.

Sequence of a specific DNA fragment of T. controversa. The inter-simple sequence repeat (ISSR) primer UBC_859 sequences are underlined with fine lines; sequence characterized amplified region (SCAR) primers ISSR140A/ISSR511A are underlined with double lines
Specificity of the SCAR marker
Of a total 75 isolates of 15 fungal species tested, all 19 TCK isolates had the expected 372 bp fragment and all isolates of the other species did not have the band (Figs. 3, 4). In a multiplex PCR test, the DNA samples of TCK, T. caries and T. laevis were successfully amplified with the ITS primers, but only the TCK isolates were amplified with the SCAR primers (Supplement Fig. 1). The results indicated that the SCAR marker is specific to TCK.

PCR amplification with sequence characterized amplified region (SCAR) primers ISSR140A/ISSR511A. Lanes 1–16, isolates TCK1–TCK16 of T. controversa; lanes 17–32 isolates, TCT1–TCT16 of T. caries; lanes 33–45, isolates TF1–TF13 of T. laevis; lane 46, negative control and M marker DL2000 fragments marked with sizes in base pair (bp). The specific band is marked with an arrow

PCR amplification with sequence characterized amplified region (SCAR) primers ISSR140A/ISSR511A. Lanes 1–3 isolates TCK17–TCK19 of T. controversa; lanes 4 and 5 isolates PS1 and PS2 of P. striiformis; lanes 6 and 7 isolates PT1 and PT2 of P. tritici; lanes 8 and 9 isolates PG1 and PG2 of P. graminis; lanes 10 and 11 isolates BG1 and BG2 of Blumaria graminis; lanes 12–14 isolates FG1–FG3 of Fusarium graminearum; lane 15 Ustilago tritici; lane 16 U. hordei; lane 17 N. horrida; lane 18 U. scitaminea; lane 19 S. sorghi; lane 20 U. maydis; lanes 21 and 22 isolates SR1 and SR2 of Sphacelotheca reiliana; and M marker DL2000 fragments marked with sizes in base pair. The specific band is marked with an arrow
Sensitivity of the SCAR marker
The sensitivity of the SCAR marker was tested using a series of dilutions of the genomic DNA extracted from isolate TCK9. The electrophoresis pattern showed that the strongest band was obtained with the highest amount of DNA (50 ng) and the band intensity decreased as the template concentration decreased (Fig. 5). The band was still visible in the PCR product amplified using 1 ng DNA template. Conceivably, 5 ng could be set as the low limit for detection of the TCK specific marker using the PCR conditions and gel electrophoresis procedures specified in this study.

Sensitivity test of different concentrations of the T. controversa DNA amplified with sequence characterized amplified region (SCAR) primers ISSR140A/ISSR511A. M marker DL2000 with fragments marked with sizes in base pair and lane 1 100 ng/μL; lane 2 50 ng/μL; lane 3 20 ng/μL; lane 4 10 ng/μL; lane 5 5 ng/μL; lane 6 1 ng/μL; lane 7 100 pg/μL; lane 8 10 pg/μL; lane 9 1 pg/μL and lane 10 0.1 pg/μL. The specific band is marked with an arrow
In addition, we were able to detect the SCAR fragment with one teliospore, as well as 2–20 spores (Fig. 6).

Teliospores of T. controversa amplified with sequence characterized amplified region (SCAR) primers ISSR140A/ISSR511A. M marker DL2000 with fragments marked with sizes in base pair and lane 1, one teliospore; lane 2, two teliospores; lane 3, three teliospores; lane 4, five teliospores; lane 5, ten teliospores; lane 6, twelve teliospores; lane 7 twenty teliospores; and lane 8 ddH2O. The specific band is marked with an arrow
Detection of wheat samples using the SCAR marker
As shown in Fig. 7, the TCK SCAR marker was detected from all samples of inoculated plants except the sample of tillering stage.

Detection of wheat samples by SCAR marker. M marker DL2000 with fragments marked with sizes in base pair and lanes 1–5, root; lanes 6–10, 1-leaf stage; lanes 11–15, 2-leaf stage; lanes 16–20, 3-leaf stage; lanes 21–25, tillering stage; lanes 26–30, jointing stage; lanes 31–35, mature stage and lanes 36–40, non-inoculated wheat plants. The specific band is marked with an arrow
Standard curve of RT-PCR
The SYBR Green I and TaqMan real-time PCRs for identifying T. controversa were developed. For SYBR Green I, ten-fold serial dilutions of plasmid DNA (CN = 2.65 × 109 − 2.65 × 104, 0.01 ng − 0.1 fg) was used as a template. The standard curve was generated as described by Yuan et al. (2009), with a linear range covering 6 log units. In Fig. 8, the correlation coefficient of the standard curve in SYBR Green I real-time PCR reached 0.995 and the amplification efficiency was 107.3 %. Furthermore, the amplification was specific as the curve melt and its detection sensitivity was 0.1 fg (CN = 2.65 × 104), showing that the SYBR Green I RT-PCR was successfully established.

Establishment of standard curve by SYBR real-time PCR. a Real-time amplified curves. 1–6 ten-fold dilutions of recombined plasmid DNA (0.01 ng–0.1 fg, CN = 2.65 × 109–2.65 × 104); 7 negative control. b Melt curve of SYBR Green I (peak temperature at 85 °C). c Standard curve
For the TaqMan probe RT-PCR, the standard curve was calculated on the serial dilutions of plasmid DNA (CN = 2.65 × 1010 − 2.65 × 104, 0.1 ng − 0.1 fg). A linear relationship was observed between the Ct values and the log concentrations of recombinant plasmid DNA. In Fig. 9, the result showed that the detection sensitivity of TaqMan PCR for TCK was 0.1 fg, equal to 2.65 × 104 CN.

Construction of standard curve by TaqMan probe real-time PCR. a Real-time amplified curves. 1–6 ten-fold dilutions of recombined plasmid DNA (0.01 ng–0.1 fg, CN = 2.65 × 109–2.65 × 104); 7 negative control. b Standard curve
Identification of TCK using TaqMan assay
The optimized TaqMan probe real-time PCR was used to detect the different TCK and T. caries isolates. In Fig. 10, the red lines are the standard curve based on diluted plasmid (0.1 ng − 0.1 fg, CN = 2.65 × 1010 − 2.65 × 104). All TCK isolates were amplified with Ct from 12 to 20 and amplified curves obtained (green lines). In contrast, the amplification of all T. caries isolates did not produce any signals, (no Ct and no amplified curves, yellow lines). The black line is the curve plot of the negative control (DNA of healthy wheat). The results confirmed that the identification of TCK with TaqMan probe real-time PCR was accurate and specific.

Detection of T. controversa and T. caries strains by TaqMan probe real-time PCR. a Real-time amplified curves. Red lines are the standard curves based on diluted plasmid (0.01 ng–0.1 fg, CN = 2.65 × 109–2.65 × 104) and the green lines represent the amplified curves of T. controversa 1–13. The yellow lines are the curve plots of T. caries and black lines are negative controls (DNA of healthy wheat). b Standard curve. (Color figure online)
Detection of wheat samples using the TaqMan assay
As shown in Fig. 11, TCK was detected from wheat samples at different growth stages using TaqMan probe real-time PCR. In contrast, no band was detected in the non-inoculated wheat samples.

Detection of T. controversa from wheat at different growth stages using the TaqMan probe real-time PCR method. a Real-time amplified curves; the red lines are the standard curves in which plasmid DNA was 2.65 × 109–2.65 × 104 CN and the green lines are the amplified curves of wheat DNA at different growth stages. The black lines are negative controls. b Standard curve. (Color figure online)
Discussion
ISSR is a molecular marker method developed for using microsatellite sequences without the requirement of genomic sequences. The primers designed for microsatellite sequences can fully scan microsatellite loci in the genome. Due to different degrees of binding of ISSR primers to the genome of different species, PCR reactions optimized for different species have low universality. We have previously attempted PCR amplification in three different ISSR reactions and found great differences in the amplified bands regarding the size and number. In the application of ISSR, the reaction should be optimized in accordance with the features of the target genome, in order to obtain a complete, stable set of polymorphic fragments. We had also attempted to amplify DNA fragments from Puccinia striiformis (the wheat stripe rust pathogen) and Blumeria graminis (the wheat powdery mildew pathogen) using PCR reactions optimized for TCK, only yielding few and blurry bands. When applying the TCK-optimized condition in amplifying T. caries and T. laevis, we obtained bands as clear and stable as those from TCK. In the present study, we identified an ISSR marker specific to TCK.
Although the ISSR marker can be used for molecular defection of TCK, many other bands are also amplified, which makes the visual examination of the TCK specific band difficult (Fig. 1). To solve the problem, we converted the ISSR marker to a SCAR marker. The SCAR marker is more stable, much easier to score and able to detect the band with a much lower amount of the template DNA. Thus, the SCAR marker is suitable for analyzing a large number of samples.
In the present study, through analysing 75 isolates of 15 fungal species including TCK and its closely related species, T. caries and T. laevis, its related species by ISSR techniques, we found that the SCAR marker keeps the specificity of the ISSR marker. Using the SCAR marker, we successfully discriminated TCK from other species. This method is significantly faster than the conventional methods used in identification and monitoring of TCK. Previously, Liu et al. (2009) developed SCAR markers using AFLP, Gao et al. (2010, 2011) developed SCAR markers also using the ISSR approach. Their markers can successfully differentiate TCK from T. caries and T. laevis. Similar to their detection limit of template DNA, the marker developed in the present study also enables the use of DANA as low as 1 ng in a 25 µl PCR mixture to detect a TCK-specific band.
In this study, we also tested the specificity of the SCAR marker using broken teliospores directly instead of extracted DNA from teliospores, which is much more flexible and saves the time needed for extracting DNA. We also used a large number of fungal species including several smut fungal species. Therefore, the marker has potential to distinguish TCK from other bunt or smut fungi produced in other crops or grasses that have potential to contaminate wheat grain.
The SYBR Green I and TaqMan real-time PCR methods were used in this study to quickly and accurately identify TCK. Both detection sensitivity of SYBR Green I and TaqMan PCRs was 0.1 fg, equal to 2.65 × 104 CN. Compared with common PCR amplification, both methods provide a simple and straightforward design. We also developed TaqMan RT-PCR to detect TCK and T. caries at the same time (Fig. 10). However, our study of SYBR Green I was more sensitive (0.1 fg) than that of Yuan et al. (2009), who reported a sensitivity of 1.0 fg.
We also detected TCK from wheat plants grown from seeds artificially contaminated by TCK teliospores at various growth stages except the tillering stage with TaqMan RT-PCR (Fig. 11), in consistent with Yuan et al. (2009). The fragment was not amplified from the tillering samples perhaps because the fungal mass might be too little. The similar result was also obtained with the SCAR primers in the regular PCR experiment (Fig. 7). This phenomenon was not mentioned in Yuan et al. (2009), but is interesting to be studied further to understand the pathogen development and distribution in various plant tissues a different growth stages.
In summary, we developed a SCAR marker and SYBR Green I and TaqMan RT-PCR approaches for quick and accurate detection of TCK. The marker and the approaches can be used to in tests not only with extracted DNA, but also with teliospores and asymptomatic wheat samples with a high sensitivity.
References
Arcade A, Anselin F, Faivre RP, Lesage M, Paques LE, Prat D (2000) Application of AFLP, RAPD and ISSR markers to genetic mapping of European and Japanese larch. Theor Appl Genet 100:299–307
Baylis RJ (1958) Studies of Tilletia controversa, the cause of dwarf bunt of wheat. Can J Bot 36:17–32
Davila JA, Lorce Y, Ferrer E (1999) Molecular characterization and genetic mapping of random applified microsatellite polymorphism in Barley. Theor Appl Genet 98:265–273
Duran R, Fishcher GW (1961) The genus Tilletia. Washington State University Press, Washington (USA)
Gao L, Chen WQ, Liu TG (2010) Development of a SCAR marker by inter-simple sequence repeat for diagnosis of dwarf bunt of wheat and detection of Tilletia controversa Kühn. Folia Microbiol 55:258–264
Gao L, Chen WQ, Liu TG (2011) An ISSR-based approach for the molecular detection and diagnosis of dwarf bunt of wheat, caused by Tilletia controversa Kühn. J Phytopathol 159:155–158
Gupta M, Chyi YS, Romero-Severson J, Owen JL (1994) Amplification of DNA markers from evolutionarily diverse genomes using single primers of simple-sequence repeats. Theor Appl Genet 89:998–1006
Hess WM, Frione EJ (1986) Use of electron microscopy to characterize teliospores of Tilletia caries and T. controversa. Plant Dis 70:458–460
Hoffmann JA (1982) Bunt of wheat. Plant Dis 66:979–986
Kochanova M, Zouhar M, Prokinova E, Rysanek P (2004) Detection of Tilletia controversa and Tilletia caries. Plant Soil Environ 50(2):75–77
Kojima T, Nagaoka T, Ogihara Y (1998) Genetic linkage map of ISSR and RAPD markers in Einkom wheat in relation to that of RFLP markers. Theor Appl Genet 96:37–45
Lian C, Zhou Z, Hogetsu T (2001) A simple method for developing microsatellite markers using amplified fragments of inter-simple sequence repeat (ISSR). J Plant Res 114:381–385
Liu JH, Gao L, Liu TG, Chen WQ (2009) Development of a sequence-characterized amplified region marker for diagnosis of dwarf bunt of wheat and detection of Tilletia controversa Kühn. Lett Appl Microbiol 49:235–240
McDonald JG, Wong E, Klassen GR (2000) Differentiation of Tilletia species by rep-PCR genomic fingerprinting. Plant Dis 84:1121–1125
Meekins JF, Ballard HE, McCarthy BC (2001) Genetic variation and molecular biogeography of a northern American invasive plant species (Alliaria petiolata, Brassicaceae). Int J Plant Sci 162:161–169
Meiners JP, Waldher JT (1959) Factors affecting spore germination of twelve species of Tilletia from cereals and grasses. Phytopathology 49:724–728
Menzies JG, Bakkeren G, Matheson F, Procunier JD, Woods S (2003) Use of inter-simple sequence repeats and amplified fragment length polymorphisms to analyze genetic relationships among small grain-infecting species of Ustilago. Phytopathology 23:167–175
Pimentel G (2000) Genetic variation among natural populations of Tilletia controversa and T. bromi. Phytopathology 90:376–383
Pimentel G, Carris LM, Levy L, Meyer RJ (1998) Genetic variability among isolates of Tilletia barclayana, T. indica and allied species. Mycologia 90:1017–1027
Scott KD, Eggler P, Seaton G, Rossetto M, Ablett EM, Lee LS, Henry RJ (2000) Analysis of SSRs derived from grape ESTs. Theor Appl Genet 100:723–726
Sun JH, Li ZC, Jewett DK, Britton KO, Ye WH, Ge XJ (2005) Genetic diversity of Pueraria lobata (Kudzu) and closely related taxa as revealed by inter-simple sequence repeat analysis. Weed Res 45:255–260
Talhinhas P, Neves-Martins J, Leitao J (2003) AFLP, ISSR and RAPD markers reveal high levels of genetic diversity among Lupinus spp. Plant Breed 122:507–510
Trione EJ (1982) Dwarf bunt of wheat and its importance in international wheat trade. Plant Dis 66:1083–1088
United States Department of Agriculture (1998) Risk assessment for the importation of US wheat containing teliospores of Tilletia controversa (TCK) into the Peoples Republic of China. United States Department of Agriculture, Agricultural Research Service and World Agricultural Outlook Board, Washington
Young PA (1935) A new variety of Tilletia tritici in Montana. Phytopathology 25:40–44
Yuan Q, Nian SJ, Yin YP, Li MH, Cai J, Wang ZK (2009) Development of a PCR-based diagnostic tool specific to wheat dwarf bunt, caused by Tilletia controversa. Eur J Plant Pathol 124:585–594
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genome 20:176–183
Zupunski V, Ignjatovic-Micic D, Nikolic A, Stankovic S, Jevtic R, Levic J, Ivanovic D (2011) Identification of Tilletia species using Rep-PCR fingerprinting technique. Genetika 43(1):183–195
Acknowledgments
We appreciate Xianming Chen (USDA-ARS and Washington State University, USA) for critical revising the paper before we submitted. We appreciate Blair J. Goates, UADA-ARS, National Small-Grains Germplasm Research Facility for providing T. controversa and T. caries. This work was supported by Beijing Natural Science Foundation (Grant No. L140012), Multidisciplinary Cooperation Project of Beijing Nova Program (Grant No. Z141105001814116), Beijing Nova Program (Grant No. Z131105000413057) and Ministry of Agriculture, P. R. China (Grant No. CARS-03).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gao, L., Yu, H., Han, W. et al. Development of a SCAR marker for molecular detection and diagnosis of Tilletia controversa Kühn, the causal fungus of wheat dwarf bunt. World J Microbiol Biotechnol 30, 3185–3195 (2014). https://doi.org/10.1007/s11274-014-1746-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1746-5