
Abstract
Rice blast, caused by Magnaporthe grisea threatens rice production worldwide. It is important to develop novel and environment-safe strategies to control the fungus. Here we reported that Bacillus subtilis KB-1122 could strikingly inhibit the growth of M. grisea P131 in agar diffusion assays. To further understand the molecular mechanism on the suppressive role of B. subtilis on M. grisea, the antagonist–pathogen interaction of the two strains was studied by using comparative proteome analysis in this report. The cellular and culture supernatant (CSN) proteins were prepared from co-culture and subjected to two-dimensional polyacrylamide gel electrophoresis. Proteome analysis revealed 33 cellular and 18 CSN proteins showing changes upon co-culture respectively. Importantly, down-regulated cellular proteins came from M. grisea, whereas up-regulated proteins derived from B. subtilis. Results suggested that glyceraldehyde-3-phosphate dehydrogenase and serine protein kinase might contribute to antifungal activity of B. subtilis KB-1122. Of CSN proteins identified, the endo-1,4-beta-glucanase (involved in degradation of polysaccharides) was up-regulated consistently at different times of incubation. This suggests that this enzyme plays an important role in the interaction between B. subtilis KB-1122 with M. grisea P131.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the important ecology questions challenging microbiologists and plant pathologists now is how to put crop diseases under the control effectively with the application of environmental-friendly approaches instead of relying on the extensive use of synthesized chemical pesticides. The use of beneficial microorganisms is considered as one of the most promising methods for more rational and crop safe-management practices (Fravel 2005). Members of the genus Bacillus are often documented as microbial factories for the production of a vast array of biologically active molecules that have potential inhibition on phytopathogen growth, such as kanosamine or zwittermycin A from Balillus cereus (Emmert and Handelsman 1999).
More recently, intensive efforts have focused on using the biocontrol agents for protecting fruit and vegetable crops from post-harvest diseases (Mari et al. 1996; Batta 2004). The action mode of most postharvest biocontrol agents was assessed using traditional screening procedures (Castoria et al. 1997). Antagonists were selected based on the demonstrated efficiency of direct interactions or the evidence that substances (or antibiotics) toxic to the potential pathogens were secreted into the growth medium. Strains of Bacillus subtilis have also been reported as biocontrol agents for controlling plant pathogens (Korsten et al. 1997). Including B. subtilis, the antifungal properties of Bacillus species were further investigated for their importance in the biological control of a number of plant and animal diseases (Grover et al. 2010; Ryder et al. 1999; Whipps 2001).
Among the fungal diseases infesting crops, the blast disease caused by the haploid, filamentous, heterothallic fungus Magnaporthe grisea is the most devastating and causes a huge loss of crop yields up to 60 % per year. Management of fungal diseases such as rice blast through the repeated application of chemical control agents leads to the development of chemical-resistant strains of pathogens. Moreover, the indiscriminate uses of chemicals have caused a serious threat to the environment and food safety (Taguchi et al. 2003). The conventional breeding method is the most economical strategy for disease resistance improvement, but this has only been partially successful because of the lack of information about the blast pathogen and its diversity. Therefore, it is very important and urgent to develop alternative strategies to control this disease and reduce the loss of crops. Literatures indicated that many effective microorganisms were isolated and their efficacy has been tested in greenhouse and small field experiments (Yang et al. 2008). There are no reports on B. subtilis isolates being used as biological control agents for the rice disease.
Among the many physiological races of rice blast pathogen, M. grisea P131, has acted as a representative pathogen used in many researches (Zhang et al. 2008a, b, 2009). We previously characterized B. subtilis strains KB-1111 and KB-1122, and demonstrated their efficient antifungal activities against indicator fungi pathogen M. grisea P131. Furthermore, we took advantage of proteomic approach to compare the cellular and extracellular protein profiles of B. subtilis KB-1111and B. subtilis KB-1122 which cultured under normal conditions. Besides, the proteins associated with the strong antagonistic ability of B. subtilis KB-1122 (Zhang et al. 2009) were identified in this study. Accordingly, in order to understand the antifungal mechanisms of B. subtilis against M. grisea pathogen, we further investigated the macromolecular proteins that mediated the interaction between B. subtilis KB-1122 and M. grisea P131.
Materials and methods
Microorganisms and growth conditions
Bacillus subtilis KB-1122 was kindly provided by Kureha Chemical Industry Co., Ltd., Japan. This strain grew aerobically in Luria–Bertani (LB) broth in shake-flasks at 28 °C and 160 rpm. Bacteria growth was monitored by measurement of the optical densities (OD) at 600 nm, when the OD600 values reached 1.0–1.2, the physiology of B. subtilis KB-1122 cells would reach to stationary phase. As an indicator pathogen strain, M. grisea P131 was kindly provided by Dr. You-Liang Peng, Department of Plant Pathology, The China Agriculture University. To generate this fungal strain for co-culture assays, M. grisea P131 mycelium was incubated on potato dextrose agar (PDA) and then allowed to grow at 28 °C for 7 days. Several pieces of fungal disks (5 mm diameter) from freshly-grown culture were transferred into LB broth, and incubated under the same culture conditions as B. subtilis KB-1122.
Co-culture assays of B. subtilis KB-1122 and M. grisea P131
Bacillus subtilis KB-1122 cells were cultured in LB broth (300 mL/L Liter flask) until OD600 values reached 1.0–1.2. Cells were harvested by centrifugation (15,000g for 15 min at 4 °C) and washed twice with sterile water. M. grisea P131 mycelium incubated in LB broth for 48 h, the mycelial pellets were collected and washed twice with sterile water. The co-culture experiments were carried out by resuspension of B. subtilis KB-1122 cells (1.5 × 108 cell/mL) and M. grisea P131 mycelial pellets (0.05 g/mL) in the same shake-flasks at 28 °C with LB broth. The co-culture samples were incubated for 3, 6, 9, 12, and 24 h in culture conditions as described previously, and the initial co-cultures without incubation were used as a control (0 h time point). All procedures described above were carried out under sterile conditions. For microscopy assay, these co-culture samples were observed under a Zeiss Axioskop 40 microscope (Carl Zeiss). Images were captured by using the Axiocam MRc digital camera (Carl Zeiss), exported as TIFF files, and adjusted with Adobe Photoshop software (Adobe, San Jose, CA, USA). All experiments were performed in three replicates.
The antifungal activity of B. subtilis KB-1122 against M. grisea P131 was determined by agar diffusion inhibition assay (Zhang et al. 2009; Bonev et al. 2008).
Extraction of cellular proteins and culture supernatant (CSN) proteins
Cellular proteins were extracted according to a published protocol (Kim et al. 2001) with slight modifications. Co-culture samples grew and were harvested at indicated time points (0, 3, 6, 9, and 12 h), and after washing thoroughly with potassium phosphate buffer (pH 7.4), the samples were centrifuged at 5,000g for 10 min at 4 °C respectively, to remove bound extracellular proteins and other debris. The co-culture pellets were lysed with 2 mL of precooled extraction buffer followed with a brief sonication (0.6 cycles, 60 % amplitude for 1 min) on ice. The homogenate was centrifuged at 15,000g for 15 min at 4 °C. The supernatant was mixed with an equal volume of Tris-buffered phenol and then vortexed for 10 min. The phenol phase was collected, and after the proteins were precipitated by addition of three volume of 100 mM ammonium acetate methanol, and the mixture was incubated overnight at −20 °C. The protein pellets were collected by centrifugation at 15,000g for 15 min at 4 °C, and subsequently washed three times with cold acetone. After centrifugation, the pellets were vacuum-dried and resuspended in rehydration buffer (1 mg protein mass in 100 μL rehydration buffer) containing 7 M urea, 2 M thiourea, 4 % CHAPS, 2 % ampholine (pH 3.5–10), and 20 mM DTT. The protein solution was used for two-dimensional electrophoresis (2-DE) or maintained at −80 °C for further use.
To extract proteins from CSN, the co-culture samples routinely grew and harvested at indicated time points (3, 6, 9, and 12 h), and the cells and mycelial pellets in the suspension were removed by filtration through 0.22-μm-pore size membrane. The proteins in the cell-free filtrate were precipitated in chilled 20 % (w/v) trichloroacetic acid (TCA) for 30 min (Nouwens et al. 2002). The precipitates were harvested by centrifugation at 15,000g for 45 min at 4 °C, washed five times with cold acetone and vacuum-dried and solubilized in rehydration buffer as described previously.
2-DE analysis
Two-dimensional electrophoresis was carried out according to the method as described by Abbasi and Komatsu (2004) with some modifications. The first dimensional isoelectric focusing (IEF) was performed in a 13-cm long glass tube with 3 mm in diameter. The gel mixture contained 4 % acrylamide, 2 % NP-40, 5 % carrier ampholytes (one part pH 3.5–10, one part pH 5–8). In general, about 600 μg of proteins from co-cultured cellular and 500 μg of proteins from CSN were loaded onto each gel, respectively. IEF was performed at 200 V for 30 min, 400 V for 15 h, and then 800 V for 1 h. After the first-dimensional separation, IEF gels were incubated in equilibration buffer for 15 min twice. The second-dimensional electrophoresis was conducted using 15 % polyacrylamide gels with 5 % stacking gels (130 × 140 × 1 mm). The gels were stained with Coomassie Brilliant Blue (CBB) R-250.
The molecular masses (Mr) of proteins on gels were determined by co-electrophoresis of standard protein markers low molecular weight calibration kit for SDS electrophoresis, molecular mass range (Mr) 14,400–97,000, GE Healthcare), and pI of the proteins were determined by migration of the protein spots on 13-cm (pH 3.5–10 and 5–8) IEF gels.
Images of CBB-stained gels were recorded by UMAX Power Look 2100XL scanner (Maxium Tech, Taipei, China) with a resolution of 300 dots per inch (dpi). The transparency mode was used to obtain a grayscale image. The image analysis was performed with ImageMaster™ 2D Platinum software. Spot detection was carried out automatically, and the optimized parameters were as follows: saliency 2.5, partial threshold 4, and minimum area 50. To minimize the variations in experiments of protein extracts, at least triplicate gels, resulting from protein extracts obtained from independent experiments, were analyzed for each sample, and there were 3-time technical repetition with the analysis sample. The identified protein spots were confirmed manually. The normalized values of protein spots on three replicate 2D gels of each treatment were exported to SPSS Version 13.0 (Lead Technologies, Chicago, Illinois, USA) for statistical analysis. Only those with significant and consistent changes were counted as differentially accumulated proteins (>1.5 fold, p < 0.05).
In-gel digestion, MS analysis, and database searching
Protein spots of candidate were excised from the gels manually and cut into small pieces. Protein digestion and MALDI–TOF-MS analysis were performed as described previously by Shen et al. (2003) with slight modifications. Briefly, each small gel piece with protein was destained with 50 mM NH4HCO3 in 50 % (v/v) methanol for 1 h at 40 °C. The destaining step was repeated until the gel was colorless. The protein in the gel piece was reduced with 10 mM DTT in 100 mM NH4HCO3 for 1 h at 60 °C and incubated with 40 mM iodoacetamide in 100 mM NH4HCO3 for 30 min at room temperature. The gel pieces were minced and lyophilized, and then rehydrated in 25 mM NH4HCO3 with 10 ng sequencing-grade modified trypsin (Promega, Madison, WI, USA) at 37 °C overnight. After digestion, the protein peptides were collected and the gel pieces were washed with 0.1 % TFA in 50 % ACN three times to collect the remaining peptides. Tryptic peptide masses were measured with a MALDI–TOF mass spectrometer (Shimadzu Biotech, Kyoto, Japan).
The National Center for Biotechnology Information (NCBI) database was searched for the acquired peptide mass fingerprinting (PMF) data using the MASCOT software available at (http://www.matrixscience.com). B. subtilis or other fungi were chosen for the taxonomic category. Searches were conducted using 0.2 Da as the mass error tolerance, and one missed cleavage site was allowed for each search. The unmentioned parameters were set according to their default values in the software. To determine the confidence of the identification results, the following criteria were used: besides the minimum MOWSE score (51 for B. subtilis; 67 for M. grisea), results showed matched queries above four and sequences coverage above 15 % were chosen as positive. Only the best matches with high confidence levels were selected.
Results
Antimicrobial effects and mycelium abnormality
As shown in Fig. 1, the growth of M. grisea P131 was apparently restrained by B. subtilis KB-1122 (Fig. 1b) as compared to control (Fig. 1a). B. subtilis KB-1122 produces inhibition zones on the spread of M. grisea P131, and the growth pattern of the pathogen mycelium exhibited the distinct suppression by KB-1122 as compared the adjacent regions with far-reached regions (Fig. 1b). This observation indicated that KB-1122 has a strong antifungal activity against M. grisea P131.

Antifungal activity of B. subtilis KB-1122 against M. grisea P131. a The normal growth of M. grisea P131 without inhibition. b Abnormal hyphal diffusion of M. grisea P131 challenged by B. subtilis KB-1122. An aliquot (6 μL) of sterile distilled water were added to each disk as a control (a). An aliquot (6 μL) culture of B. subtilis strains in log phase were added to each disk, respectively (b)
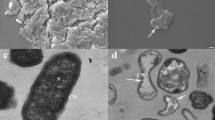
To obtain a better understanding of the intrinsic molecular changes in the interaction, we conducted a co-culture assay with these two strains of microorganisms. As demonstrated in the microscopic photographs of confronting culture (Fig. 2), the abnormal mycelium growth of M. grisea P131 was observed when co-cultured with B. subtilis KB-1122. After incubation for 3 h, B. subtilis KB-1122 cells showed a strong swarming motility, and the formation of communities enclosing the mycelia of M. grisea P131. Meanwhile, the initial irregular mycelia were observed (Fig. 2, 3 h, arrow indicated). After 9 h, the distorted mycelia ramification and abnormal germ tubes were evidently noticeable (Fig. 2). After incubation for 24 h, the number of the integrated mycelium of M. grisea P131 decreased significantly and less mycelial debris suspended in the co-culture solution. The results indicated that B. subtilis KB-1122 inhibited the growth of M. grisea P131 through the mycelium hydrolysis or degradation with secreted biological compounds.

Microscopic images on structure changes of the mycelium of M. grisea P131 in the time course of interaction with B. subtilis KB-1122 cells. Arrows indicate the abnormal mycelia. Bars = 5 μm
Comparison of proteome patterns in the interaction
Based on the microscopic observation on the interaction (Fig. 2), we reasoned that proteomic analysis would allow us to identify the induced alterations of proteins for the interaction between B. subtilis KB-1122 and M. grisea P131. To do so, the co-cultured samples incubated at the intervals of 3, 6, 9 and 12 h were prepared and subjected to 2-DE electrophoresis. The initial co-cultures without incubation (0 h time point) were used as control. To ensure the reproducible results, the experiments were repeated three times. After CBB R-250 staining and ImageMaster 2D Platinum software analysis, general proteome patterns in the range of pH 3.5–10 were generated for both intracellular proteins from cellular preparation (Fig. 3) and extracellular proteins from CSN (Fig. 4). The results revealed that at each time point, approximate 800 protein spots were detected from the samples of cellular proteins (Fig. 3), while up to 200 protein spots were found in CSN (Fig. 4).

Expression patterns of differentially regulated cellular proteins in the interaction process of B. subtilis KB-1122 and M. grisea P131. a Expression profiles of the initial cocultures without incubation (0 h time point). b Co-cultured samples incubated for 9 h. c Enlarged images to show quantitative changes of differentially regulated cellular proteins expressed at indicated times. d Shows quantitative changes of these proteins expressed at indicated times. Data is representatives of three independent experiments and shown as intensity mean ± SD. The protein spots are numbered, corresponding to the numbers in Table 1. Arrows indicate the changed proteins. U stands for the up-regulated protein; D stands for the down regulated protein

Expression patterns of differentially regulated culture supernatant (CSN) proteins in the interaction process of B. subtilis KB-1122 and M. grisea P131. a Expression profiles of the initial cocultures without incubation (0 h). b After co-culture incubation for 9 h. c Enlarged images to show quantitative changes of culture supernatant (CSN) proteins. d Shows quantitative changes of these CSN proteins expressed at indicated times. Data is representatives of three independent experiments and shown as intensity mean ± SD. The protein spots are numbered, corresponding to the numbers in Table 2. Arrows indicate the changed proteins, CU up-regulated CSN protein; CD down regulated CSN protein
We then investigated the molecular alterations of intracellular proteins by performing comparative analysis using cellular fractions at each time point of incubation. With a 1.5-fold quantitative change as the criteria, 39 protein spots were found to show their differential expressions in the cellular fractions upon incubation for 3, 6, 9 and 12 h. In order to distinguish the dynamic statuses of individual interesting proteins, we marked each protein spot with a number (Fig. 3). As demonstrated in Fig. 3c, enlarged images revealed that the levels of some interesting proteins, as reflected by the imaging density, indeed displayed dramatic changes in a time-dependent manner. Among them, 21 proteins were down regulated and 18 proteins were up regulated (arrows indicated). All of these down regulated proteins decreased their levels markedly in the beginning period of incubation (3 h), and their levels decreased more with elongated incubation times (6, 9 and 12 h) (Fig. 3c, d). Several protein spots became almost undetectable at 12 h time point as compared to that at 0 h control (Fig. 3, spots D1, D10 and others). For the up-regulated proteins, about 50 % of interested protein spots increased their levels strikingly in the first 3 h of incubation. After that, their levels of proteins increased slightly (spots U8 and U14), remained the same level (spot U4 and U17) or decreased slightly (spots U15) under elongated incubation times (Fig. 3). The significance of these changes on some interesting proteins was summarized in the histogram chart (Fig. 3d).
In order to characterize the CSN proteins in response to the interaction, CSN proteins were extracted from the co-culture media after incubation with 3, 6, 9, and 12 h, and then subjected to 2-DE. We originally pursued to obtain the secretory protein profile of M. grisea P131 as the control by growing the fungus up to 5 days for CSN protein extraction. However, the routine methods as described previously failed to precipitate enough CSN proteins for 2-DE analysis. This notion indicated that M. grisea P131 secreted very little of CSN proteins at the early phase of co-culture incubation. Due to lack of the extracellular proteome of M. grisea P131 used as the control, we decided to generate the secretory protein profile of B. subtilis KB-1122 from the medium with overnight incubation, and then used as the control to compare the differential CSN proteins expressed in the time course of interaction. As a result, more than 200 CSN protein spots with a M r between 14 and 90 kDa and a pI ranging from pH 3.5 to 10 were detected on gel of each treatment (Fig. 4). The CSN protein profiles were very similar to the secretory protein profiles expressed by B. subtilis KB-1122. Of 16 changed proteins, seven proteins were down regulated and nine proteins were up regulated (Fig. 4).
Identification of cellular proteins differentially expressed in the time course of interaction
As shown in figures, 33 out of 39 cellular proteins displaying the dramatic alterations (Fig. 3) were identified with Mowse scores greater than the threshold (Table 1). Among these individual spots, the abundance of 21 cellular proteins was decreased, while that of 12 cellular proteins was increased. Based on the identification results, about 21 down-regulated cellular proteins were highly matched with M. grisea species, and the 12 up-regulated proteins were from B. subtilis. These protein changes may lead to the inhibitory growth and degradation of integrated mycelium in M. grisea P131 in co-culture incubation (Fig. 2).
These identified proteins were classified into three groups based on their functions: (1) protein synthesis, (2) protein modification and signal transduction, and (3) metabolisms of carbohydrate and energy (Table 1). The largest group contained the proteins involved in carbohydrate metabolism and energy production, such as 4 down-regulated proteins (Fig. 3c, spots D11, D12, D15, and D18) and 6 up-regulated proteins (Fig. 3c, spots U7, U9, and U14–17). Interestingly, 2 protein spots (spots D18 from M. grisea and U15 from B. subtilis) were annotated as the same protein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which plays a crucial role in the glycolysis pathway (Hughes et al. 2002; Zhou et al. 2006). Notably, we found a serine protein kinase (spot U8), which was involved in the regulation of bacterial signal transduction (Macek et al. 2007), and increased gradually in the process of fungal-bacterial interaction. In addition, several enzymes (spots U4–U6, and U10) involved in the protein synthesis (Bigot et al. 2006; Endo and Kurusu 2007), increased greatly during the process of co-culture incubation. Moreover, in the time course of interaction, we found that most of these identified cellular protein spots showed dramatically changes (up or down) at a particular time point, 3 h, except for spot U8 (Fig. 3), indicating that the essential biochemical changes occurred in both strains at the first 3 h during the interaction process.
Identification of culture supernatant (CSN) proteins differentially expressed in the time course of interaction
We applied a same strategy to identify the altered proteins in CSN and to understand their contributions to essential pathways in the interaction. We identified 14 differentially expressed proteins in CSN preparation, in which one protein spot was annotated as a hypothetical protein from M. grisea and the others were from B. subtilis species (Table 2). These identified CSN proteins were also classified into three main groups: (1) energy production and conversion (spots CD2–3, CU2 and CU4), (2) hydrolysis-related enzymes (spots CU1, CD4–7 and CU6–7), and (3) cell motility and secretion (spot CU3). Interestingly, three groups for CSN proteins were different from that for cellular proteins. As indicated in Table 2, 7 out of 14 identified CSN proteins were enzymes for hydrolysis-related functions. Among these hydrolytic enzymes, two proteins were annotated as the same phytase (spots CU1 and CD4) that involves the hydrolysis of phytic acid and other organophosphate substrates (Kerovuo et al. 2000). One protein spot was identified as endo-1,4-beta-xylanase (spot CD7) that hydrolyzes the xylan into xylose and xylo-oligosaccharides (Chu et al. 2000). In addition, three protein spots (spots CD5–6, CU7) were identified as neutral protease precursors (Table 2) that may act as potential virulence factors in defense response (Huang et al. 2005; Tian et al. 2007). Their levels were decreased gradually in the time course of co-culture incubation. It is noteworthy that one protein spot (spot CU6), which was identified as endo-1,4-beta-glucanase and involved in the degradation of fungal cell wall (Kim et al. 2004), was up-regulated in the first 9 h. Furthermore, 4 proteins (spots CD2–3, CU2 and CU4) involved in energy pathway and one flagellin (spot CU3) involved in cell motility (Titz et al. 2006) were also identified in CSN proteins.
Discussion
In this study, the interaction between B. subtilis KB-1122 and M. grisea P131 was performed through a co-culture approach. Combined with the antifungal capacity of antagonistic strain, 2-DE technique was used to obtain the global analysis of cellular and CSN proteins differentially expressed in the time course of co-culture incubation. These results provide us valuable insights into understanding molecular mechanisms occurring in this interaction between these two strains.
The inhibition effects of B. subtilis KB-1122 on M. grisea P131
Our study suggests that the inhibitory activities of B. subtilis strain KB-1122 will show potential benefits to crops and environments. It can be used as effective bio-control agents for many crop diseases, especially for the worldwide destructive pathogen of rice, M. grisea (Zhang et al. 2008a, 2009). Convincing evidence indicates that the co-culture method may exactly mimic a real biological circumstance of antagonist-pathogen interaction, and has been considered as an adequate communicative response in antifungal assays (Benhamou and Chet 1997; Boonchan et al. 2000). In our co-culture assays of B. subtilis KB-1122 and M. grisea P131, the abnormal mycelial growth of M. grisea P131 was observed at different time points of incubation (Fig. 2).
The results indicated that B. subtilis KB-1122 may secrete molecules that mediate the interaction as early as 3 h after incubation. Some extracellular signaling molecules may mediate quorum sensing, such as the formation of communities enclosing the mycelia of M. grisea P131, and may also play important roles in suppressing some metabolic pathways in M. grisea P131. Results from 2-DE analysis provide pivotal biochemical evidence on the morphological changes and degradation of mycelia observed under microscopy.
Differentially expressed cellular proteins specific for the antagonistic activity of B. subtilis KB-1122
To characterize the cellular proteins in response to the interaction, the differentially expressed cellular proteins were classified into several categories. Among these differentially expressed proteins, the most notable one (spots D18 from M. grisea and U15 from B. subtilis) was glyceraldehyde-3-phosphate dehydrogenase (GAPDH) that catalyzes a reversible reaction involved in glycolysis process. GAPDH was also postulated the characterization of a particular virulence factor with a promoting role in the pathogenic bacteria infection on the host (Hughes et al. 2002). In the interaction between Fusarium graminearum and wheat, GAPDH was also differentially upregulated in both F. graminearum and wheat (Zhou et al. 2006). Our results suggested that GAPDH might contribute to the antifungal activities of antagonistic B. subtilis KB-1122. Serine protein kinase (spot U8) is in the family of serine/threonine/tyrosine kinases that participate in protein phosphorylation of B. subtilis cells (Macek et al. 2007). Activation of this kinase played a critical role in regulation of signal transduction involved in mediation of bacterial virulence (Macek et al. 2007). The up-regulation of this protein suggested that the enzyme might be correlated to the regulation of its self-defense in B. subtilis, which resulted in a repression of GAPDH induction in targeted pathogens M. grisea (Echenique et al. 2004).
A large portion of function cellular proteins in this study was involved in carbohydrate metabolism and energy production. In addition to the GAPDH (spots D18 and U15) mentioned previously, seven other proteins [2 from M. grisea (spots D11 and D15) and 5 from B. subtilis (spots U7, U9, U14, and U16–17)] were found to be critical enzymes involved in glycolysis and tricarboxylic acid (TCA) cycles. Among these, three enzymes (spots D11, D15 and D18) involved in glycolysis pathway, were dramatically decreased in the first 3 h of incubation. Meanwhile, six other up-regulated cellular proteins involved in glycolysis and TCA cycles, were significantly increased in co-culture incubation (Fig. 3). The decreased activity of three enzymes (from M. grisea) may lead to hindrance of the whole metabolism pathway (Fig. 5a). On the other hand, the increased activity of six enzymes (from B. subtilis) might result in activation of glycolysis and TCA cycle (Fig. 5a). These results indicated that, during the course of interaction, the whole carbohydrate metabolism pathway was activated in B. subtilis KB-1122 but repressed in M. grisea P131. These observations suggest that the differentially expressed enzymes involved in carbohydrate metabolism pathway may determine the antagonistic activity of B. subtilis KB-1122 against M. grisea P131 in the interaction (Fig. 5b).

Scheme of metabolic pathways and possible antifungal mechanism in the interaction of B. subtilis KB-1122 against M. grisea P131. a Shows the differentially regulated proteins involved in glycolysis and tricarboxylic acid (TCA) cycles. b Shows the possible antifungal mechanism
Hydrolytic enzymes in CSN enhance the antagonistic activity of B. subtilis KB-1122 against M. grisea P131
Recent studies on CSN proteome of pathogenic bacteria revealed that some CSN proteins may be critical for the virulence of bacterial pathogen (Mattow et al. 2003). With the advantage of proteomic technology, it is necessary to identify the changes of important hydrolytic enzymes in CSN proteins and further understand the possible antifungal mechanism of B. subtilis KB-1122 against M. grisea P131. In this study, among those identified CSN proteins, 7 out of 14 were annotated as hydrolytic enzymes belonging to B. subtilis species (Table 2). The hydrolytic enzymes showed the complicated patterns of changes, indicating that multiple pathways may be regulated in this interaction.
Previous studies suggested that microbial extracellular proteases in some antagonist strains could act as virulence antagonism factor involved in hydrolysis of fungal cell walls (Almeida et al. 2007). Endo-1,4-beta-glucanase may play a function role in the degradation of polysaccharides, and directly involve in antifungal defense responses (Kim et al. 2004; Levy et al. 2002). In this study, we found that endo-1,4-beta-glucanase (spot CU6) was consistently up-regulated in the time course of co-culture incubation, suggesting that this enzyme may play a decisive role in this interaction of B. subtilis KB-1122 against M. grisea P131. However, we are still puzzled about the exact reasons of upregulation of endo-1,4-beta-glucanase.
The synergistic action of cellular and CSN proteins in the interaction between B. subtilis and M. grisea
In the present study, we investigated the molecular mechanisms of the fungal-bacterial interaction using a comparative proteome approach. Our results land a support that GAPDH, serine protein kinase and endo-1,4-beta-glucanase identified in cellular and CSN proteome may play important roles in the antifungal function of B. subtilis KB-1122. Serine protein kinase (spot U8) upregulated in KB-1122 as essential signal transduction molecules may promote the activities of hydrolytic enzymes, virulence factor and carbohydrate metabolism pathways when KB-1122 fights against P131 (Fig. 5). In addition, GAPDH, a unique virulence factor, was upregulated in KB-1122 (U15) but down-regulated in P131, indicated that different levels of this factor may contribute to the noticeable antagonistic activity of KB-1122 against P131. Most importantly, the differentially expressed cellular proteins may initiate the antagonistic activity of KB-1122 by secreting antifungal hydrolytic enzymes into the co-culture environments. The sustained activities of these hydrolytic enzymes in CSN, including endo-1,4-beta-glucanase (spot CU6), certainly permit a continuous hydrolysis of the cell wall of pathogen (Fig. 5). To this end, this synergistic action between cellular and CSN proteins efficiently represses the viability and growth of pathogen P131. Yet, due to the complexity of the biology involved in the interaction between B. subtilis KB-1122 and M. grisea P131, it will be also of interest to define key factors contributing to the changes of identified proteins. Therefore, further investigation will warrant more valuable insights onto the elucidation of molecular mechanisms on suppression of M. grisea.
Overall, our study identified that proteomic profiles in cellular and CSN protein from B. subtilis and M. grisea pathogen specific to their interaction. The antifungal mechanisms of B. subtilis against M. grisea pathogen may be contributed by the synergistic action of cellular and CSN proteins. Our findings also provide the valuable information for further studies on the application of B. subtilis KB-1122 isolates as biological agents to control rice blast or other crop diseases.
Abbreviations
- LB:
-
Luria–Bertani broth
- PDA:
-
Potato dextrose agar
- CSN:
-
Culture supernatant
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
References
Abbasi FM, Komatsu S (2004) A proteomic approach to analyze salt-responsive proteins in rice leaf sheath. Proteomics 4:2072–2081
Almeida FBR, Cerqueira FM, Nascimento Silva RN, Ulhoa CJ, Lima AL (2007) Mycoparasitism studies of Trichoderma harzianum strains against Rhizoctonia solani: evaluation of coiling and hydrolytic enzyme production. Biotechnol Lett 29:1189–1193
Batta YA (2004) Postharvest biological control of apple gray mold by Trichoderma harzianum Rifai formulated in an invert emulsion. Crop Prot 23:19–26
Benhamou N, Chet I (1997) Cellular and molecular mechanisms involved in the interaction between Trichoderma harzianum and Pythium ultimum. Appl Environ Microbiol 63:2095–2099
Bigot A, Botton E, Dubail I, Charbit A (2006) A homolog of Bacillus subtilis trigger factor in Listeria monocytogenes is involved in stress tolerance and bacterial virulence. Appl Environ Microbol 72:6623–6631
Bonev B, Hooper J, Parisot J (2008) Principles of assessing bacterial susceptibility to antibiotics using the agar diffusion method. J Antimicrob Chemother 61:1295–1301
Boonchan S, Britz ML, Stanley GA (2000) Degradation and mineralization of high-molecular-weight polycyclic aromatic hydrocarbons by defined fungal–bacterial cocultures. Appl Environ Microbiol 66:1007–1019
Castoria R, De Curtis F, Lima G, Cicco VD (1997) β-1,3-Glucanase activity of two saprophytic yeasts and possible mode of action against postharvest diseases. Postharvest Biol Technol 12:293–300
Chu PW, Yap MN, Wu CY, Huang CM, Pan FM (2000) A proteomic analysis of secreted proteins from xylan-induced Bacllius sp. strain K-1. Electrophoresis 21:1740–1745
Echenique J, Kadioglu A, Romao S, Andrew PW, Trombe MC (2004) Protein Serine/Threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect Immun 72:2434–2437
Emmert EAB, Handelsman J (1999) Biocontrol of plant disease: a (Gram+) positive perspective. FEMS Microbiol Lett 171:1–9
Endo A, Kurusu Y (2007) Identification of in vivo substrates of the chaperonin GroEL from Bacillus subtilis. Biosci Biotechnol Biochem 71:1073–1077
Fravel DR (2005) Commercialization and implementation of biocontrol. Annu Rev Phytopathol 43:337–359
Grover M, Nain L, Singh SB, Saxena AK (2010) Molecular and biochemical approaches for characterization of antifungal trait of a potent biocontrol agent Bacillus subtilis RP24. Curr Microbiol 60:99–106
Huang XW, Tian B, Niu Q, Yang J, Zhang L (2005) An extracellular protease from Brevibacillus laterosporus G4 without parasporal crystals can serve as a pathogenic factor in infection of nematodes. Res Microbiol 156:719–727
Hughes MJG, Moore JC, Lane JD, Wilson R (2002) Identification of major outer surface proteins of Streptococcus agalactiae. Infect Immun 70:1254–1259
Kerovuo J, Lappalainen I, Reinikainen T (2000) The metal dependence of Bacillus subtilis phytase. Biochem Biophys Res Commun 268:365–369
Kim ST, Cho KS, Jang YS (2001) Two-dimensional electrophoretic analysis of rice proteins by polyethylene glycol fractionation for protein arrays. Electrophoresis 22:2103–2109
Kim KH, Kim YO, Ko BS, Youn HJ, Lee DS (2004) Over-expression of the gene (bglBC1) from Bacillus circulans encoding an endo-β-(1–3), (1–4)-glucanase useful for the preparation of oligosaccharides from barley β-glucan. Biotechnol Lett 26:1749–1755
Korsten L, De Villiers EE, Wehner FC (1997) Field sprays of Bacillus subtilis and fungicides for control of preharvest fruit diseases of avocado in South Africa. Plant Dis 81:455–459
Levy I, Shani Z, Shoseyocv O (2002) Modification of polysaccharides and plant cell wall by endo-1,4-β-glucanase and cellulose-binding domains. Biomol Eng 19:17–30
Macek B, Mijakovic I, Olsen JV, Gnad F (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol Cell Proteomics 6:697–707
Mari M, Guizzardi M, Brunelli M, Folchi A (1996) Postharvest biological control of grey mold (Botrytis cinerea) on fresh-market tomatoes with Bacillus amyloliquefaciens. Crop Prot 15:699–705
Mattow J, Schaible UE, Schmidt F, Hagens K (2003) Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24:3405–3420
Nouwens AS, Willcox MDP, Walsh BJ (2002) Proteomic comparison of membrane and extracellular proteins from invasive (PAO1) and cytotoxic (6206) strains of Pseudomonas aeruginosa. Proteomics 2:1325–1346
Ryder MH, Yan Z, Terrace TE, Rovira AD (1999) Use of strains of Bacillus isolated in China to suppress take-all and rhizoctonia root rot, and promote seedling growth of glasshouse grown wheat in Australian soils. Soil Biol Biochem 31:19–29
Shen SH, Jing YX, Kuang TY (2003) Proteomics approach to identify wound-response related proteins from rice leaf sheath. Proteomics 3:527–535
Taguchi Y, Hyakumachi M, Horinouchi H (2003) Biological control of rice blast disease by Bacillus subtilis IK-1080. Ann Phytopathol Soc Jpn 69:85–93
Tian BY, Yang JK, Lian LH, Wang C, Li N (2007) Role of an extracellular neutral protease in infection against nematodes by Brevibacillus laterosporus strain G4. Appl Microbiol Biotechnol 74:372–380
Titz B, Rajagopala SV, Ester C, Hauser R (2006) Novel conserved assembly factor of the bacterial flagellum. J Bacteriol 188:7700–7706
Whipps JM (2001) Microbial interactions and biocontrol in the rhizosphere. J Exp Bot 52:487–511
Yang JH, Liu HX, Zhu GM, Pan YL (2008) Diversity analysis of antagonists from rice-associated bacteria and their application in biocontrol of rice diseases. J Appl Microbiol 104:91–104
Zhang CX, Zhao X, Jing YX, Chida T, Chen H (2008a) Phenotypic and biological properties of two antagonist Bacillus subtilis strains. World J Microbiol Biotechnol 24:2179–2181
Zhang J, Peng YL, Guo ZJ (2008b) Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res 18:508–521
Zhang CX, Zhao X, Han F, Yang MF (2009) Comparative proteome analysis of two antagonist Bacillus subtilis strains. J Microbiol Biotechnol 19(4):351–357
Zhou WC, Eudes F, Laroche A (2006) Identification of differentially regulated proteins in response to a compatible interaction between the pathogen Fusarium graminearum and its host, Triticum aestivum. Proteomics 6:4599–4609
Acknowledgments
This work was supported by the State Key Basic Research and Development Plan of China (2010CB126503), China Agriculture Research System (CARS-28), and the National Natural Science Foundation of China (30900968).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, C., Zhang, X. & Shen, S. Proteome analysis for antifungal effects of Bacillus subtilis KB-1122 on Magnaporthe grisea P131. World J Microbiol Biotechnol 30, 1763–1774 (2014). https://doi.org/10.1007/s11274-014-1596-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1596-1