
Abstract
Extracellular cellulase free xylanase from Thermomyces lanuginosus sp. SS-8, isolated from self heating plant wreckage material was identified as β-1,4-endo-xylanase precursor, a monomer of 21.3 kDa with no carbohydrate residue. This xylanase retained 80 % activity at 60 °C for 96 h, was active at a wide pH range of 3–11 and uniquely hydrolyzed xylan to xylose without production of xylo-oligosaccharides. Gene xynSS8 encoding xylanase from T. lanuginosus SS-8 was cloned and functionally expressed in Escherichia coli XL1 Blue using pTZ57R/T plasmid and xynSS8/pQE-9 expression vector construct respectively. Gene xynSS8 was of 777 bp and deduced amino acid sequence was a mature xylanase of 258 amino acids. XynSS8 has extra 33 amino acids compared to its nearest homolog and was thermo-alkali tolerant as that of native protein. The xylanase could degrade pulp and release substantial chromophoric materials and lignin derived compounds indicating its effective utility in pulp bleaching. Novel characteristics of the enzyme may contribute to its wide industrial usage. This is first report of cloning and functional expression of the novel xylanase from T. lanuginosus SS-8.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Xylan, a complex plant structural polysaccharide with various functional groups is second most abundant polysaccharide in nature (Prade 1995). It is important for fiber cohesion and plant cell wall integrity (Beg et al. 2001), found in large quantities in hardwoods, softwoods as well as in annual plant (Singh et al. 2003) and is located in the secondary as well as primary cell wall of plants.
Xylanases are glycosidase which catalyze the hydrolysis of 1,4-β-d-xylosidic linkages in xylan. Complete hydrolysis of xylan requires a large variety of cooperatively acting enzymes (Biely 1985). Endo-1,4-β-d-xylanases randomly cleave the xylan backbone, β-d-xylosidases cleave xylose monomers from the non-reducing end of xylo-oligosaccharides and xylobiose while removal of the side groups is catalyzed by α-l-arabinofuranosidases, α-d-glucuronidases, acetylxylan esterases etc. Xylotriose has been reported to be the smallest xylo-oligosaccharide obtained during hydrolysis of xylan by xylanases (Ryan et al. 2003) and even if some xylanases liberate xylose during hydrolysis, it accompanied with higher oligosaccharides. A current survey on xylanases discusses its distribution, structural aspects, biochemistry, applications etc. in details (Sharma and Kumar 2013).
Thermostable enzymes are increasingly in demand as resistance to thermal inactivation is a desirable property for many industrial applications (Singh et al. 2000). Major applications of thermostable xylanase are in pulp and paper industries, fruit and vegetable processing, brewing, wine production, baking, animal feeds, starch, textiles industries, fruit softening, seed germination, plant defense system, etc. (Prade 1995; Beg et al. 2001).
Thermomyces lanuginosus (formerly known as Humicola lanuginosa) is an imperfect fungus. It is noncellulytic, septate, reproduces asexually by forming aleurioconidia (Hudson 1992). Some strains of this Ascomycetes fungi have been reported earlier to produce cellulase-free thermostable β-xylanases (Singh et al. 2003; Gaffney et al. 2009; Shrivastava et al. 2009). Purification and partial characterization of the novel xylanase and its unique mode of action has been reported by us earlier (Shrivastava et al. 2011).
In the present study we report complete characterization, cloning and expression of the novel xylanases from T. lanuginosus SS-8 in Escherichia coli. Purified extracellular cellulase free novel xylanase from an indigenous strain T. lanuginosus SS-8 was identified and completely characterized. The gene encoding xylanase xynSS8 was cloned and functionally expressed in E. coli. Recombinant xylanase was purified and partially characterized suggesting that deduced amino acid sequence of XynSS8 belonged to family 11 xylanase.
Materials and methods
Fungal and bacterial strains, xylanase production, enzyme assay, protein estimation, plasmids and primers
Thermophilic xylan degrading fungi taxonomically identified as T. lanuginosus SS-8 is a potential xylanase producer (Shrivastava et al. 2011). The fungal xylanase was produced and purified as described by Shrivastava et al. (2008, 2011). Xylanase activity was assayed by method described by Miller with slight modification (Miller 1959) and protein concentration was determined by method of Bradford (Bradford 1976) using bovine serum albumin as standard.
Details of bacterial strain, plasmids and primers have been given in Table 1. The plasmid pTZ57R/T was precleaved with Eco321 and treated with terminal deoxynucleotydyl transferase at the multiple cloning sites. E. coli was grown on Luria–Bertani medium at 37 °C for 12–18 h.
Identification of XynSS8 protein from T. lanuginosus SS-8
Identification of the protein XynSS8 from T. lanuginosus SS-8 was done by analysis of mass spectra of tryptic peptide digest using ESI SYNAPT HDMS (Waters Corporation, Milford, MA, USA).
Purified protein was resolved on 18 % SDS-PAGE gel, stained with Coomassie Brilliant Blue R-250, and digested in-gel with trypsin using the Trypsin Profile IGD Kit (Sigma Aldrich CO, St. Louis, MO, USA), as per manufacturer’s instruction. Vacuum dried purified protein, dissolved in 200 μl of 3 % acetonitrile was injected into an online Nanoacquity ESI SYNAPT HDMS fitted with a Nanotrap desalting column (Symmetry C18, particle size 5 μm, 180 μm × 20 mm) and a Nano column (BEH 130 C18, particle size 1.7 μm, 75 μm × 150 mm) and was analyzed at a flow rate of 500 nl min−1 on solvent A (0.1 % formic acid in water) and 3–50 % gradient of solvent B (0.1 % formic acid in acetonitrile) in 30 min.
Glufibrinopeptide (GFP) solution was delivered through reference sprayer of the NanoLockSpray source and the MS/MS fragment ions (mass range of 100–1,600 m/z) of GFP were used to obtain the final mass spectrometer calibration. Analysis was done on nanoESI positive mode with source temperature 80 °C. Capillary voltage was maintained at 3.5 kV, cone voltage 35 V, extraction voltage 4 V and gas flow at cone was maintained at 50 l h−1. Accurate mass data were collected using 6 and 15–40 eV of trap cell for MS and MSE data acquisition respectively (Silva et al. 2005). Data acquisition was done in the presence of lock mass (Leu Enk) to get accurate mass measurement.
Chromatograms obtained were analyzed with MassLynx software version 4.1 (Waters Corporation, Manchester, UK) and peptide mass fingerprinting search for the MS/MS spectra was performed at MASCOT (www.matrixscience.com). Raw mass spectra data was processed and submitted to ProteinLynx Global Server (PLGS) v 2.3 (Waters Corporation, Milford, MA, USA) for identification of the protein using default parameters. Database search parameters were as follows: enzyme trypsin, allowing up to one missed cleavage, fixed modification: carbamidomethyl (C), variable modification: deamidated (NQ), taxonomy selected for search was fungi, 0.2 Da peptide tolerance was allowed and query protein mass was selected as 21 kDa.
Characterization of xylanase
Purity of eluted proteins was checked through electrophoresis after each purification step on native (Ornstein and Davis 1964) and SDS-PAGE (Laemlli 1970) and all gels were visualized after silver staining (Blum et al. 1987). Zymogram was obtained by using an agarose overlay gel prepared by mixing 1.5 % OSX (10 ml) with 2 % agarose in 50 mM citrate buffer, pH 6.5 (20 ml) (Royer and Nakas 1990). Purified protein, after electrophoresis on native gels, was overlaid with the substrate agarose gel and incubated at 50 °C for 30 min. Substrate gel was immersed in 95 % ethanol for 60 min to reveal the enzyme-active bands. Isoelectric point (pI) of the enzyme was determined on immobilized pH gradient (IPG) strips (7 cm, pH 3–10 and 3–6) using a Protean IEF Cell (both from Biorad Laboratories, Hercules, CA, USA) following the procedure suggested by the manufacturer. Total carbohydrate content was determined by phenol–sulphuric acid method (Dubois et al. 1956) with glucose as standard.
Optimal temperature and pH of purified xylanase was determined by performing standard activity assays at temperatures from 4 °C to 100 °C and pH 3 to 11. In all cases, enzyme and substrates were pre-incubated separately at the selected temperature for 15 min and then mixed and further incubated for 10 min at that temperature. Buffers used for study were 50 mM sodium acetate buffer (pH 3–6), Tris–Cl buffer (pH 7–8) and Glycine–NaOH buffer (pH 9–11). Thermal stability up to 7 days was determined by performing assays after incubating the enzyme at 50, 60 and 70 °C at pH values that showed maximum activity at selected temperature. The pH stability of enzyme was assessed at all pH values from 3 to 11 at 60 °C for period up to 7 days.
Hydrolytic activities of xylanase against natural and synthetic substrates like beechwood xylan (BWX), OSX, CMC was assayed. Substrates (20 mg ml−1) dissolved in 50 mM sodium acetate buffer, pH 6.0 were incubated with purified xylanase for 10 min at 60 °C, and amounts of reducing sugar released were estimated. Kinetic parameters (Michaelis–Menten constant, Km and maximal reaction velocity, Vmax) were calculated at optimal assay conditions using different concentrations (2–40 mg ml−1) of OSX and BWX by linear regression from double reciprocal plot according to Lineweaver and Burk (1934).
Effects of salts KCl, NaCl, CaCl2, MnSO4, MgSO4, FeSO4, CuSO4 and HgCl2 (1 and 10 mM concentration) and reagents like dithiothreitol (DTT), ethylene diamine tetra acetic acid (EDTA), sodium dodecyl sulphate (SDS), nonyl phenoxylpolyethoxylethanol (NP-40) and ß-mercaptoethanol (1 and 10 mM or 1 and 5 % concentration) that modify amino acids and proteins were determined by performing assay with OSX in 50 mM sodium acetate buffer pH 6.0 as substrate. Residual activity was expressed as the percentage of activity observed in absence of any compound.
Treatment of pulp by xylanase and determination of lignin derived compounds and chromophoric materials
Bagasse pulp was treated with xylanase following protocol described by Bissoon et al. (2002), with slight modification. Pulp was thoroughly washed with distilled water for obtaining neutral pH of wash water. Pulp equivalent to 10 g dry weight and 10 % consistency was charged with 5, 50, 150 U purified xylanase per gram pulp and incubated in polyethylene bags at 60 °C for 3 h with intermittent kneading. Pulp slurry was filtered and pulp was thoroughly washed with water. Control samples were treated under same condition with inactivated enzymes. Effect of xylanase on pulp samples was observed by scanning electron microscopy (JEOL JSM-6390, Japan). Ddehydrated pulp samples were frozen and vacuum-dried for 48 h. Specimens were coated with gold–palladium and were analyzed through secondary electron imaging at 10 kV accelerating voltage. Enzyme mediated release of lignin derived compounds and chromophoric materials from pulp samples were measured at 280 and 465 nm respectively.
Isolation of total RNA, cDNA synthesis and cloning
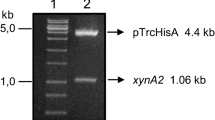
RNA was extracted from T. lanuginosus SS-8 mycelia induced for xylanase production according to the protocols suggested by Zhou et al. (2008) with minor modifications and was reverse transcribed to cDNA by RevertAid Premium Enzyme Mix (RevertAid First Strand cDNA Synthesis Kit; PureExtreme; Fermentas Life Sciences, Fermentas UAB, Lithuania) following manufacturer’s instruction. Gene xynSS8 was amplified with XynF and XynR primers using Taq DNA polymerase (Fermentas GmbH, St. Leon-Kot, Germany) under following conditions: denaturation at 95 °C for 2 min followed by 40 cycles of denaturation at 94 °C for 20 s, annealing at 60 °C for 1 min and polymerization at 72 °C for 2 min and final extension at 72 °C for 30 min. PCR amplified gel purified xynSS8 was ligated to pTZ57R/T and xynSS8/pTZ57R/T was transformed into E.coli XL1 Blue cells by TA cloning method using reagents and following instructions as in InsTAclone PCR Cloning procedure (InsTAclone PCR Cloning kit, Fermentas Life Sciences, Fermentas UAB, Lithuania). Plasmids from amprLacZ− clone (white colonies) were analyzed for presence of xynSS8, nucleotide sequence of insert was obtained by primer walking at the sequencing services facility of Bangalore Genei (Bangalore Genei, Bangalore, India) and open reading frame (ORF) was analyzed using NCBI sequence analysis (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Homology search was performed using GenBank BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and multiple sequence alignments were carried out with ClustalW2 program (http://www.ebi.ac.uk/Tools/clustalw2/).
Functional expression of xynSS8 in E. coli
Escherichia coli XL1 Blue was used to express β-1,4-endo-xylanase as N-terminal 6× His tag construct under control of T5 promoter of expression vector pQE-9 (Qiagen GmbH, Quiagen, Strasse, Germany). Plasmid DNA was isolated from recombinant E. coli XL1 Blue containing xynSS8/pTZ57R/T. Gene xynSS8 in pTZ57R/T was amplified with XynRS-F and XynRS-R primers using Taq DNA polymerase (Fermentas GmbH, St. Leon-Kot, Germany) following parameters: denaturation at 95 °C for 2 min followed by 25 cycles of denaturation at 94 °C for 20 s, annealing at 60 °C for 1 min and polymerization at 72 °C for 2 min and final extension at 72 °C for 10 min. Xylanase gene fragment xynSS8 was digested with Bam HI and Hind III and ligated in frame with N-terminal His-tags peptide sequence in pQE-9 vector. Expression vector construct xynSS8/pQE-9 were transformed into E. coli XL1 Blue by heat shock method following manufacturer’s instruction (The Quiaexpressionist 2003) and were selected on LB agar supplemented with ampicillin (100 μg/ml) and kanamycin (25 μg/ml) at 37 °C for 24 h. Positive transformants were screened on LB agar with 1 mM IPTG (Isopropyl-β-d-thiogalactopyranoside) and 100 μg/ml of ampicillin and transformed colonies were analyzed for IPTG induced expression of xynSS8 gene.
Purification and partial characterization of recombinant xylanase
Transformed E. coli were grown on LB medium supplemented with ampicillin (100 μg/ml) and induced with 1 mM IPTG to express recombinant xylanase, XynSS8. Induced cells (10 ml) were harvested by centrifuging at 15,000×g for 1 min, resuspended in 200 μl of lysis buffer (50 mM NaH2PO4; 300 mM NaCl; 10 mM immidazole, pH 8.0) and lysed with 1 mg ml−1 lysozyme. Disrupted cells were centrifuged and clear cell supernatant was purified by Ni2+ immobilized metal ion affinity chromatography (IMAC) on agarose by batch purification method (The Quiexpressionist, Quiagen GmbH, Strasse, Germany) following manufacturer’s instruction. Purity of active fraction was confirmed by SDS-PAGE.
Temperature tolerance of the recombinant xylanase was determined by incubating it with 2 % substrate (in buffer, pH 6.0) at temperatures 50, 60, 70, 80 and 90 °C for 10 min and estimating the reducing sugar released by dinitrosalycylic acid. pH tolerance of recombinant xylanase was determined at pH 6.0, 7.0, 8.0 and 9.0 by incubating enzyme with 2 % substrate for 10 min at 60 °C and estimating the reducing sugar released after reaction by dinitrosalicylic acid (Miller 1959).
Results
Protein identification
Xylanase from T. lanuginosus SS-8 was purified to homogeneity (Shrivastava et al. 2011) and identified. Masses of intact peptides and fragmentation of all peptides were obtained from data generated using low trap energy and elevated energy, respectively. Molecular weight of the purified protein as obtained by nanoLC ESI MS spectra was 21.3 kDa. The nanoLC ESI MS MSE data of the protein digest was submitted to PLGS v 2.3 and identified as endo-β-1,4-xylanase precursor using MASCOT (http://www.matrixscience.com). Derived peptide sequences AGLNVNGDHYYQIVATEGYFSSGYAR (2,853.3115 MH+, Fig. 1), GWNPGLNAR (985.485 MH+), ITVADVG (674.371 MH+) and VNAPSIDGTQTFDQYWSVR (2184.035 MH+) have been submitted to Uniprot (Uniprot accession code O43097).

ESI MS/MS spectra of β-1,4-endo-xylanase precursor. The sequence is AGLNVNGDHYYQIVATEGYFSSGYAR
Characterization of xylanase from T. lanuginosus SS-8
Positive zymogram was obtained on native polyacrylamide gel. Purified enzyme was identified as monomer of 23.79 kDa (ImageQuant 1D gel analysis software), having pI 3.9 without carbohydrate residue.
Optimum temperature for enzyme activity varied with different pH values (Fig. 2). Purified xylanase was thermostable, highly active at temperatures between 50 and 70 °C and showed activity at wide pH range from 3 to 11 (Fig. 3, data for pH 3 and pH 4 not shown). It retained more than 80 % of its activity even after 96 h of incubation at 60 °C at pH 6.0, hydrolyzed OSX and BWX and was completely inactive on CMC. It followed Michaelis–Menten relationship on OSX as well BWX at pH 6 and 60 °C and hydrolyzed OSX more efficiently than BWX. The apparent Km and Vmax values for OSX and BWX were 10.87, 17.67 mg ml−1 and 1,818.2, 2,222.2 U mg−1, respectively. Turnover number (Kcat value) of for hydrolyzing OSX and BWX were 2.6 × 103 and 3.43 × 103 s−1 respectively. Xylanase required 3.85 × 10−4 and 2.92 × 10−4 s to perform each catalyzed reaction for hydrolyzing OSX and BWX, respectively.

Effect of pH and temperature on activity of purified xylanase incubated with OSX for 15 min. Hundred percent xylanase activity is 218.6 Units/ml

Xylanase stability at varied pH range at 60 °C. Hundred percent xylanase activity is 218.6 Units/ml
Monovalent cations, K+ and Na+ slightly stimulated the enzyme activity. Divalent cations showed mixed results, Ca2+ and Mn2+ promoted, Mg2+ had no effect and Fe2+, Cu2+ and Hg2+ strongly inhibited enzyme activity. Disulphide reducing agents, β-mercaptoethanol and DTT strongly activated the enzyme. EDTA had no effect on the enzyme activity, SDS strongly inhibited while NP40 strongly promoted the enzyme activity.
Effect of xylanase treatment on pulp
Scanning electron imaging showed that pulp fibers treated with inactivated enzyme were uniform, intact and smooth in appearance without fibril formation. On the contrary, surface of xylanase treated pulp was rough and striated with minor cracks and pores (Fig. 4). This will increase accessibility of xylanase treated lignin for chemicals, showing bleach boosting effect. Release of lignin derived compounds and chromophoric materials were considerably higher in xylanase treated pulp samples.

Scanning electron imaging a Bagasse pulp treated with inactivated xylanase, magnification ×5,000. b Bagasse pulp treated with active xylanase, magnification ×2,500
Cloning, sequencing and analysis of xylanase gene from T. lanuginosus SS-8
Escherichia coli XL1 Blue strains were transformed with an efficiency of 7.1 × 103 transformants per μg of xynSS8/pTZ57R/T DNA. The positive transformants, i.e. amprLacZ− clones had 800 bp insert. Transformed cells had ORF consisting of 777 bp coding a xylanase gene belonging to family 11, which had 57 % G + C content with a preference of C in third position of codon.
Primary structure of XynSS8 from T. lanuginosus SS8
Gene xynSS8 encodes a polypeptide of 258 amino acids (GenBank Accession number: AEH57194.1). Deduced amino acid sequence showed a calculated molecular mass of 28,041 Da and a pI of 5.37. SignalP 3.0 program demonstrated that XynSS8 included a signal peptide of 20 amino acids and a probable mature xylanase of 238 amino acids. Comparison of the xylanase gene (xynSS8; genbank accession number JF919610) from T. lanuginosus SS-8 on homology search showed that xynSS8 was highly similar to T. lanuginosus (GU166389), T. lanuginosus (U35436), Paecilomyces sp. J18 (FJ593404), Helminthosporium turcicum (AJ238895), etc. (Fig. 5). This xylanase has extra 33 amino acids as compared to xylanase of T. lanuginosus (Schlacher et al. 1996). Two totally conserved Glutamate residues of xylanase which correspond to residues E-150 and E-242 in T. lanuginosus SS-8 xylanase have been reported to be involved in enzymatic activities (Wakarchuk et al. 1994), low molecular weight and low pI are typical characteristics of family 11 xylanases (Torronen and Rouvinen 1997).

Alignment of XynSS-8 catalytic domain with similar xylanases. Amino acid number begins with start codon. AEH5719 = T. lanuginosus SS-8, XynSS8; AAB94633 = T. lanuginosus XynA; ACY69861 = T. lanuginosus XynA; ACS26244 = Paecilomyces sp. J18, XynA; CAB52471 = Helminthosporium turcicum Xyl1. Amino acid 92–124 are extra 33 amino acids in XynSS-8 as compared to other xylanases
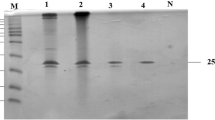
Expression and purification of recombinant xylanase in E. coli
Gene xynA was expressed under control of T5 promoter of pQE-9 expression vector regulated by the repressible lac operon. Transformed cells induced with IPTG produced recombinant xylanase XynSS8. The expressed protein was present in soluble as well as insoluble fraction as inclusion bodies and showed xylanase activity. Single band of about 23 kDa corresponding to molecular mass of mature xylanase appeared after purification on IMAC (Fig. 6). Molecular weight of N-terminal Histag fusion peptide XynSS8 expressed in E. coli was about 3 kDa greater than the native protein as obvious with other recombinant enzymes (Schlacher et al. 1996; Li et al. 2006; Wang et al. 2007; Zhang et al. 2010). Recombinant xylanase was thermostable (active at temperatures 50, 60 and 70 °C) and similar to the native enzyme it showed minor activity at 80 °C, becoming inactive at 90 °C. It was active at pH 6.0–8.0 and lost its complete activity at pH 9.0.

Immobilized metal affinity chromatography for purification of recombinant protein. Lane 1 protein low molecular weight marker (PMWL, Bangalore Genei, India); lane 2 non induced cells; lane 3 induced cell; lane 4: flow through; lane 5: wash; lane 6: purified recombinant protein
Discussion
Most industrially used xylanases are from mesophilic fungi (Deng et al. 2006; Zhou et al. 2008) which do not cater to all industrial demands, and there is continuous demand of more efficient thermostable xylanases. Our isolate, T. lanuginosus SS-8 produced thermo tolerant xylanase, active at wide pH range as compared to other fungal xylanases (Li et al. 2006; Zhou et al. 2008; Sriyapai et al. 2011; Hung et al. 2011; Do et al. 2012). This enzyme could effectively hydrolyze xylan to monomer unit without producing any oligosaccharide unit (Shrivastava et al. 2011). Thermal stability of few xylanases has been enhanced by introducing disulphide bridge (Wang et al. 2012).
XynSS8 has extra 33 amino acids compared to its nearest homolog, which could probably be a reason behind its unique mode of action. XynSS8 was successfully expressed in E. coli and the recombinant protein retained thermostability. Most recombinant xylanases reported are not thermotolerant (Ruanglek et al. 2007) or lose activity at around 65 °C or lower temperature (Xue et al. 1995; Sriyapai et al. 2011; Verma and Satyanarayana 2012).
XynSS8 is able to produce excess of xylose on hydrolysis of xylan which could prove effective in various xylose utilizing industries. Major application may include bio-ethanol and xylitol production and its unique mode of action will cater major functions alone, dismissing requirement of enzyme cocktails (Ichinose et al. 2011). Study on varied application of XynSS8 is ongoing.
References
Beg QK, Kapoor M, Mahajan L, Hoondal GS (2001) Microbial xylanases and their industrial applications: a review. Appl Microbiol Biotechnol 56:326–338
Biely P (1985) Microbial xylanolytic systems. Trends Biotechnol 3:286–290
Bissoon S, Christov L, Singh S (2002) Bleach boosting effects of purified xylanase from Thermomyces lanuginosus SSBP on bagasse pulp. Process Biochem 37:567–572
Blum H, Beier H, Gross B (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8:93–99
Bradford MM (1976) A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Deng P, Li D, Cao Y, Lu W, Wang C (2006) Cloning of a gene encoding an acidophilic endo-β-1,4-xylanase obtained from Aspergillus niger CGMCC1067 and constitutive expression in Pichia pastoris. Enzyme Microb Technol 39:1096–1102
Do TT, Quyen DT, Dam TH (2012) Purification and characterization of an acid-stable and organic solvent-tolerant xylanase from Aspergillus awamori VTCC-F312. ScienceAsia 38:157–165
Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F (1956) Colorimetric method for determination of sugars and related substances. Anal Chem 58:350–356
Gaffney M, Carberry S, Doyle S, Murphy R (2009) Purification and characterisation of a xylanase from Thermomyces lanuginosus and its functional expression by Pichia pastoris. Enzyme Microb Technol 45:348–354
Hudson HJ (1992) Fungal biology. Cambridge University Press, Cambridge, pp 106–170
Hung KS, Liu SM, Tzou WS, Lin FP, Pan CL, Fang TY, Sun KH, Tang SJ (2011) Characterization of a novel GH10 thermostable, halophilic xylanase from the marine bacterium Thermoanaerobacterium saccharolyticum NTOU1. Process Biochem 46:1257–1263
Ichinose H, Diertavitian S, Fujimoto Z, Kuno A, Leggio LL, Kaneko S (2011) Structure-based engineering of glucose specificity in a family 10 xylanase from Streptomyces olivaceoviridis E-86. Process Biochem 47:358–365
Laemlli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Li L, Tian H, Cheng Y, Jiang Z, Yang S (2006) Purification and characterization of a thermostable cellulase-free xylanase from the newly isolated Paecilomyces themophila. Enzyme Microb Technol 38:780–787
Lineweaver H, Burk D (1934) The determination of the enzyme dissociation. J Am Chem Soc 56:658–666
Miller GH (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–429
Ornstein L, Davis BJ (1964) Disc electrophoresis. I. Background and theory. Ann NY Acad Sci 121:321–349
Prade RA (1995) Xylanases: from biology to biotechnology. Biotechnol Genet Eng Rev 13:101–131
Royer JC, Nakas JP (1990) Simple, sensitive zymogram technique for detection of xylanase activity in polyacrylamide gels. Appl Environ Microbiol 56:1516–1517
Ruanglek V, Sriprang R, Ratanaphan N, Tirawongsaroj P, Chantasigh D, Tanapongpipat S, Pootanakit K, Eurwilaichitr L (2007) Cloning, expression, characterization, and high cell-density production of recombinant endo-1,4-β-xylanase from Aspergillus niger in Pichia pastoris. Enzyme Microb Technol 41:19–25
Ryan SE, Nolan K, Thompson R, Gubitz GM, Savage AV, Tuohy MG (2003) Purification and characterization of a new low molecular weight endoxylanase from Penicillium capsulatum. Enzyme Microb Technol 33:775–785
Schlacher A, Holzmann K, Hayn M, Steiner W, Schwab H (1996) Cloning and characterization of the gene for the thermostable xylanase XynA from Thermomyces lanuginosus. J Biotechnol 49:211–218
Sharma M, Kumar A (2013) Xylanases: an overview. Br Biotechnol J 3:1–28
Shrivastava S, Shukla P, Mukhopadhayay K (2008) Correlative characterization of changes in hyphal morphology during xylanase production in submerged culture by Thermomyces lanuginosus SS-8. Internet J Microbiol 4:2
Shrivastava S, Poddar R, Shukla P, Mukhopadhyay K (2009) Study of codon bias perspective of fungal xylanase gene by multivariate analysis. Bioinformation 3:425–429
Shrivastava S, Shukla P, Mukhopadhyay K (2011) Purification and preliminary characterization of xylanase from Thermomyces lanuginosus strain SS-8. 3 Biotech 1:255–259
Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ et al (2005) Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem 77:2187–2200
Singh S, Pillay B, Prior BA (2000) Thermal stability of β-xylanases produced by different Thermomyces lanuginosus strains. Enzyme Microb Technol 26:502–508
Singh S, Mandala AM, Prior BA (2003) Thermomyces lanuginosus: properties of strains and their hemicellulases. FEMS Microbiol Rev 27:3–16
Sriyapai T, Somyoonsap P, Matsui K, Kawai F, Chansiri K (2011) Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in Escherichia coli and Pichia pastoris. J Biosci Bioeng 111:528–536
The Quiaexpressionist (2003) A handbook of high-level expression and purification of 6× His-tagged proteins, 5th edn. Quiagen, Valencia, pp 39–40
Torronen A, Rouvinen J (1997) Structural and functional properties of low molecular weight endo-1,4-β-xylanases. J Biotechnol 57:137–149
Verma D, Satyanarayana T (2012) Cloning, expression and applicability of thermo-alkali-stable xylanase of Geobacillus thermoleovorans in generating xylooligosaccharides from agro-residues. Bioresour Technol 107:333–338
Wakarchuk WW, Campbell RL, Sung WL, Davoodi J, Yaguchi M (1994) Mutational and crystallographic analyses of the active-site residues of Bacillus cirlulans xyalanase. Protein Sci 3:467–475
Wang YR, Zhang HL, He YZ, Luo HY, Yao B (2007) Characterization, gene cloning, and expression of a novel xylanase XYNB from Streptomyces olivaceoviridis A1. Aquaculture 267:328–334
Wang Y, Fu Z, Huang H, Zhang H, Yao B, Xiong H et al (2012) Improved thermal performance of Thermomyces lanuginosus GH11 xylanase by engineering of an Nterminal disulfide bridge. Bioresour Technol 112:275–279
Xue GP, Denman SE, Glassop D, Johnson JS, Dierens LM, Gobius KS, Anyward JH (1995) Modification of a xylanase cDNA isolated from an anaerobic fungus Neocallimastix patriciarum for high-level expression in Escherichia coli. J Biotechnol 38:269–277
Zhang C, Jia L, Wanga S, Qu J, Li K, Xu L, Shi Y, Yan Y (2010) Biodegradation of β-cypermethrin by two Serratia sp. with different cell surface hydrophobicity. Bioresour Technol 101:3423–3429
Zhou C, Li D, Wu M, Wang W (2008) Optimized expression of an acid xylanase from Aspergillus usamii in Pichia pastoris and its biochemical characterization. World J Microbiol Biotechnol 24:1393–1401
Acknowledgments
The authors gratefully acknowledge the Government of Jharkhand, Department of Agriculture for providing infrastructure development fund to Department of Biotechnology, Birla Institute of Technology, Mesra, Ranchi for financial supports. Smriti Shrivastava is thankful to Council of Scientific and Industrial Research for Senior Research Fellowship (Grant No: 09/554(0017)/2008 EMR-I). Authors also acknowledge Central Instrumentation Facility at Birla Institute of Technology, Mesra, India for providing Scanning electron microscopy facility and Shalini Jalan and Aarushi Sharma (B. Tech Biotechnology students at Birla institute of Technology, Mesra, Ranchi, Jharkhand, Batch 2007–2011) for performing SEM analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shrivastava, S., Shukla, P., Deepalakshmi, P.D. et al. Characterization, cloning and functional expression of novel xylanase from Thermomyces lanuginosus SS-8 isolated from self-heating plant wreckage material. World J Microbiol Biotechnol 29, 2407–2415 (2013). https://doi.org/10.1007/s11274-013-1409-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-013-1409-y