
Abstract
The gene encoding an endo-β-1,4-xylanase from an Indonesian indigenous Bacillus licheniformis strain I5 was amplified using PCR, cloned, and expressed in Escherichia coli. The nucleotide sequence of a 642 bp DNA fragment was determined, revealing one open reading frame that encoded a xylanase. Based on the nucleotide sequence, calculated molecular mass of the enzyme was 23 kDa. This xylanase has a predicted typical putative signal peptide; however, in E. coli, the active protein was located mainly in intracellular form. Neither culture supernatant of recombinant E. coli nor periplasmic fraction has significantly detectable xylanase activity. The deduced amino acid of the gene has 91% identity with that of Bacillus subtilis endoxylanase. Optimal activity of the recombinant enzyme was at pH 7 and 50°C
Similar content being viewed by others
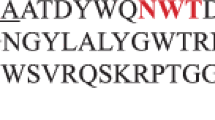


Avoid common mistakes on your manuscript.
Introduction
As a rich biodiversity country, Indonesia has many potential habitats for microbes and their industrial enzymes (http://www.apex-environmental.com/AreaDescription.html; http://www.dgtl.esdm.go.id; http://www.irgltd.com/Resources/Publications). However, for industrial needs, Indonesia has imported most of the industrial enzymes (BPPT 2006). In addition, agricultural and paper wastes are also abundantly available which sometimes can cause the environmental problems (Wiloso et al. 1995), that actually they can be converted into more valuable product using biocatalyst or enzymes. Thus, research related to isolation of Indonesian indigenous microbes which produce potential industrial enzymes and the production of these enzymes using local resources, would be very useful in Indonesia and has significant meaning to solve environmental problems.
One of the most promising industrial enzymes is xylanase (β-1,4-Endoxylanase; EC 3.2.1.8). The enzyme catalyzes the cleavage of xylan backbone at 1–4 carbon linkage to produce xylose and xylo-oligosaccharides. Xylanase is an important enzyme from biotechnological point of view, both alone or in combination with other enzymes, due to xylan as the second major abundant polymer in the world. Xylanase is one of the xylanolitic enzymes which work synergistically with other enzymes to convert this abundant polymer wastes into useful substance such as art paper, xylose, low-calorie sweetener (xylitol), or bioethanol. Xylanase is also promising enzyme to reduce the use of environmentally harmful chemical in pulp and paper industry, and predicted as a potential biocatalyst in deinking process for recycling of used paper (Kulkarni et al. 1999; Turner et al. 2007).
A large number of xylanases from different microorganisms have been described (Kulkarni et al. 1999; Subramaniyan and Prema 2002). Based on sequence similarities, the enzymes are mostly classified into two families of glycosyl hydrolases, family 10 and family 11 (Henrissat 1991). Endoxylanases belong to glycosyl hydrolase family 10 usually have larger molecular mass (more than 40 kDa); whereas those belong to glycosyl hydrolase family 11 have smaller molecular mass around 20 kDa. The family 10 xylanase usually has also detectable cellulase activity besides the xylanase activity; whereas the family 11 has not. The small size and compact folding of family 11 xylanases make it easily penetrate into the cellulose fiber network without destructing the fiber; thus, these enzymes are suitable in biobleaching (Oakley et al. 2003; Torronen and Rouvinen 1997).
We have successfully isolated some xylanolitic bacteria from Indonesia local habitats, mainly from local hot springs. One of these potential bacteria which produce xylanase has already been identified as B. licheniformis strain I5. The bacterium was isolated from Ciseeng Hot spring, West Java, Indonesia. In this paper, we described cloning and expression of family 11 xylanase gene (xyn11) of this strain in E. coli. We also reported the comparison of this recombinant xylanase with other xylanases and partial characterization of crude extract of the recombinant enzyme.
Materials and methods
Bacterial strains, plasmids, and medium
Bacterial strains which were used as hosts for plasmid amplification and expression were E. coli DH5α (Bandung Institute of Technology (ITB); genotypes: F -endA1 hsdR17 (rk - mk +) supE44 thi1 recA1 gyrA(Nalr) relA1Δ (lacZYA-argF) U169 (Ø80lacZΔM15)) and E. coli Top10 (Invitrogen; genotypes: F-mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 deoR recA1 araD139 (Δ(araA-leu)7697 galU galK rpsL endA1 nupG), respectively. Plasmids used in this experiment were pGEM-T easy (Promega, USA) and pBAD/gIII C expression vector (Invitrogen, USA). The 1.5% (w/v) agar Luria-Bertani (LB) plate mediums containing 100 μg/ml (w/v) ampicillin, 50 μg/ml (w/v) 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), and 1 mM isopropyl-β-thiogalactopyranoside (IPTG), were used for selection of E. coli harbouring recombinant plasmid. LB agar medium containing ampicillin and 0.7% (w/v) oat spelt xylan was used for confirmation of the gene expression.
Morphological and biochemical characterizations of the I5 isolate
A bacterial strain I5 which gave a clear zone around the colony on the xylan-LB medium was isolated from Ciseeng Hot spring, West Java, Indonesia. The morphological characteristics of the isolate were observed including colony and cell shape, Gram, and spore staining.
The analysis of biochemical characteristics of isolate I5 included catalase activity, urease reaction, nitrate reduction, starch hydrolysis, indole hydrolysis, carbohydrate fermentation, and gas formation.
16S rRNA gene amplification, cloning, and sequencing
Chromosomal DNA of the I5 isolate was extracted using phenol/chloroform extraction method (Sambrook et al. 2001). The genetic experiment protocols were done based on standard protocols (Sambrook et al. 2001). Amplification by PCR with universal primers 9F (5′-AGAGTTTGATC(C/A)TGGCTCAG-3′) and 1510R (5′GTTAC(G/C)TTGTTACGACTT-3′) for eubacterial rRNA gene was performed using platinum Taq DNA polymerase (Invitrogen, USA) under the following conditions. After initial 2-min hot start incubation at 94°C, the mixture was introduced to 30 cycles, each cycle including 30 s at 94°C, 45 s at 51°C, and 2 min at 72°C using a Thermal cycler (Eppendorf, Germany). A final extension was performed at 72°C for 10 min. The amplified 16S rRNA gene was purified using a commercial DNA/PCR product purification kit (Promega, USA), incubated at 72°C for 30 min to add extra adenines (A) at the 3′-end of the PCR products, and then ligated to pGEM-T easy vector at 4°C over night. The ligation mixture was used to transform E. coli DH5α. The white/blue screening with X-gal/ IPTG on LB agar medium was conducted. The positive clones then were further verified using restriction enzymes and the confirmed clones were sequenced and analyzed. The recombinant plasmids were extracted and sequenced (3130 DNA Analyzer; Applied Biosystems, USA) using T7 terminator and SP6 promoter primers with Big Dye Terminator sequence kit (Applied Biosystems). The partial 16S rRNA gene sequence was compared to known bacterial sequences in the NCBI GenBank using BLAST program. Then, the DNA sequence and the analysis result were submitted to GenBank.
Xylanase gene amplification, cloning, and sequencing
The primers for isolation xylanase gene were designed and synthesized based on the alignment of xyn11 from Bacillus retrieved from Genbank. They were 5′-AAT GCG GCC GC A ATG TTT AAG TTT AAA AAG AAT TTC T-3′ as a forward primer and 5′-GC TCT AGA TTA CCA CAC TGT TAC GTT AGA ACT T-3′ as a reverse primer. The italic and underlined sequences showed the restriction enzyme Not I site (forward primer) and EcoR I site (reverse primer) for subcloning into pBAD/gIII C expression vector. PCR amplification using the above primers and a template of extracted chromosomal DNA with Platinum Taq polymerase was performed under the following condition. Initial 94°C for 3 min for hot start, then 30 cycles of 94°C 45 s, 52°C 1 min, 72°C 2 min for each cycle The PCR product with the predicted size then was extracted and purified from the agarose gel with Gene Clean kit (Q-Biogene, USA). The next protocols were performed similarly to the protocols of the cloning and sequencing of 16S rRNA gene.
Subcloning of xylanase gene into expression vector
The inserted xyn11 with confirmed sequence was removed from the recombinant pGEM-T easy plasmid by digesting with Not I and EcoR I. Then it was subcloned into pBAD/gIII C vector at the same restriction enzymes sites. The ligation was transformed into E. coli DH5α, and the positive clone harbouring recombinant pBAD/gIII C plasmid was chosen. This recombinant plasmid then was transformed again into E. coli TOP10 using electroporation (Gene pulser, Bio-rad, USA), and the positive colony with clear zone was checked in oat spelt xylan-LB agar medium to observe the xylanase activity by the xylan–Congo red clearance plate assay. Transformation of E. coli TOP10 with empty pBAD/gIII C plasmid using electroporation was also conducted as a negative control to confirm the clear zone of the colony of the transformant.
Bioinformatics analysis of xylanase gene
The DNA sequences encoding 16S rRNA and xylanase were analyzed using CHROMAS and Genetyx software. The search for similarity of nucleotides and amino acid level was performed using BLAST program at http://www.ncbi.nlm.nih.gov. The genes then were submitted to GenBank, USA. The multiple sequences analysis was done through server http://www.genome.ad.jp. The xylanase genes and their translated amino acids which were used in multiple alignment analysis were as follows: Bsub: family 11 xylanase gene from B. subtilis (Huang et al. 2005); Bcirc: that from Bacillus circulans: (accession number AAM08360); Thermxyl: that from Thermobacillus xylanilyticus (accession number CAJ87325); Bhal: that from Bacillus halodurans (Martinez et al. 2005); Bfir: that from Bacillus firmus (Chang et al. 2004, accession number AAQ83579); Thermofu: that from Thermobifida fusca xylanase (accession number AAV64879). Signal peptide was predicted using http://www.cbs.dtu.dk server.
Recombinant enzymes preparation and enzyme assay
The colony of E. coli Top10 which bringing positive recombinant xylanase was cultivated in 50 ml LB containing ampicillin at 37°C, and at OD600 = 0.6 arabinose with concentration 0.02% (w/v) was added and the cultivation was continued for 4 h. The negative controls E. coli TOP10 without plasmid and recombinant E. coli Top10 with empty pBAD/gIII C were treated with the same manner to confirm that the xylanase activity come from the presence of recombinant plasmid pBAD/gIII C-xyn11. The supernatant of the culture was stored at 4°C for further analysis. The periplasmic protein was obtained after treating the cell pellet with osmotic shock solution (20 mM Tris–HCl pH 8, 2.5 mM EDTA, 20% (w/v) Sucrose) based on the protocol in pBAD/gIII C manual. The cytoplasmic fraction was obtained as follows: The cells were pelleted by centrifugation and suspended in 5 ml Phosphate Buffer pH 7 containing 1 mM mercaptoethanol, and disrupted by sonication (Heat systemXL Ultrasonicator, Japan) at the maximum frequency for 30 s on and 30 s off repeatedly for 10 times at 4°C. The crude extract containing the recombinant enzyme was recovered by centrifugation.
Xylanase activity was measured (each sample in duplicates) based on Miller method using dinitrosalicylic acid to detect reducing sugar, and d-xylose was used as a standard (Miller 1959; Bailey et al. 1992). Ten μl crude enzyme extract was mixed with 490 μl of 1% oat spelt xylan in 50 mM buffer at indicated pH. The enzyme mixture was then incubated at indicated temperature for 5 min, DNS reagent (1% dinitrosalicylic acid, 0.2% phenol, 0.05% sodium sulfite, and 1% sodium hydroxide, 20% (w/v) potassium sodium tartrate) 750 μl was added to stop the reaction, boiled at 100°C for 5 min, and incubated at room temperature, then added with 250 μl water and centrifuged to obtain a clear supernatant. A blank for each sample at indicated pH and temperature was the same mixture composition as that of sample, but 10 μl enzymes was added following addition of DNS into the reaction mixture. The absorbance was measured at 540 nm. The activities of periplasmic fraction and the supernatant were also measured with the exactly same procedure, however the sample volume was 50 μl. One unit xylanase activity was defined as the amount of enzyme to produce 1 μmol of xylose per min under the assay conditions.
The effect of temperature and pH on the enzyme activity and the thermal stability
The effect of the temperature on enzyme activity was measured in the temperature range of 30–80°C at pH 7 using 50 mM sodium phosphate buffer. The effect of pH on the activity was measured at 50°C within pH range of 5–11 using 50 mM of the following buffer: citrate buffer (for pH 5, 6), sodium phosphate buffer (for pH 6–8), Tris–HCl buffer (for pH 8–10), and Glysin–NaOH buffer (pH 10, 11). Thermostability of the enzyme was measured at 50°C using 50 mM sodium phosphate pH 7 after preincubating the enzyme at 40, 50, and 55°C for 10, 20, and 30 min. Protein concentration was measured by means of dye-binding assay method of Bradford, and bovine serum albumin (BSA) was used as the standard protein (Bradford 1976).
Results and discussion
In this paper, we described the isolation of a gene encoding family 11 xylanase (xyn11) from Indonesian indigenous xylanase-producing bacterium using PCR-cloning approach. This Indonesian indigenous bacterial strain was isolated from Ciseeng Hot Spring, West Java, Indonesia. The isolated strain gave the clear zone around the colony on the xylan-containing LB agar medium. The supernatant of the culture of this bacterium in LB-xylan medium had activity around 2 U/mg at 50°C. This strain also showed its ability to produce xylanase at solid state fermentation using low cost substrate Banana peels (Mahyudin et al. 2007).
Morphological and biochemical determination of this bacterial strain showed that this isolate was rod shape, motile, endospore forming and Gram positive microorganism, positive for catalase, nitrate reduction, urease, starch hydrolysis, and carbohydrate fermentation, but negative for indole production and H2S formation. Therefore, it was concluded that it belonged to genus of Bacillus based on Bergey’s manual of determinative bacteriology (Holt et al. 1994). In order to confirm the identity of this strain down to species level, the partial 16S rRNA gene sequence was determined and submitted to GenBank with accession number EU030266. The comparison with other 16S rRNA gene at GenBank revealed that the strain had 99–100% identity with B. licheniformis. Therefore, the strain I5 was considered as one strain of B. licheniformis species.
The primers for isolation of xylanase gene used in this experiment were designed based on the alignment of several family 11 endoxylanases gene from Bacillus species. All aligned sequences showed high degree of nucleotide identity, therefore the specific primers could be designed. The primers contained EcoR I and Not I restriction enzymes sites to facilitate the subcloning of this gene into pBAD/gIII C expression vector. The amplification of the chromosomal DNA of this strain using the above primers produced a specific band with similar size to the predicted size of 662 bp (including the primers). The PCR product then was ligated into pGEM-T easy TA cloning vector and transformed into E. coli DH5α. The plasmid of the E. coli clones with the correct size of insert was then sequenced. This insert with confirmed sequence was then subcloned into pBAD/gIII C vector at EcoR I and Not I sites to express the gene in E. coli. We observed there was clear zone around the positive E. coli TOP10 clone harbouring recombinant pBAD/g III C-xyn11 on the agar LB medium containing 0.7% xylan by the xylan–Congo red clearance plate assay (Fig. 1a).

Comparison of E. coli colonies using xylan–Congo red clearance protocol at agar LB medium containing 0.7% (w/v) of oat spelt xylan. (a) An E. coli TOP10 clone harbouring recombinant pBAD/gIII C plasmid containing gene of family 11 xylanase (xyn11). It exhibited xylanase activity with clear zone around the colony. (b) An E. coli TOP10 colony without plasmid. (c) An E. coli TOP10 colony with empty pBAD/gIII C plasmid
The DNA sequence is containing an ORF of 642 bp nucleotide encoding 213 amino acids with an initiation and a stop codon without frameshift. The gene obtained in this work has been submitted into GenBank with accession number DQ520129. The GC content of the gene was 41.09%. Based on the translated amino acids, the calculated molecular mass including the signal peptide was 23 kDa and isoelectric point (pI) was at pH 9.96. The deduced amino acid of the gene had also putative signal peptide as predicted by signal peptide prediction server (http://www.cbs.dtu.dk/services/SignalP/). The signal peptide was predicted located at the first 30 amino acids in this the N-terminal region (Fig. 2).

Alignment of B licheniformis I5 xylanase’s deduced amino acid studied in this work with other family 11 endoxylanases. Blich: xylanase gene from B. licheniformis (accession number DQ 520129; this study); Bsub: that from B. subtilis (Huang et al. 2005); Bcir: that from Bacillus circulans: (accession number AAM08360); Thermxyl: that from Thermobacillus xylanilyticus (accession number CAJ87325); Bhal: that from Bacillus halodurans (Martinez et al. 2005); Bfir: that from Bacillus firmus (accession number AAQ83579); Thermofu: that from Thermobifida fusca (accession number AAV 64879). Numbering starts at the N termini of the enzymes. The bold and underlined amino acid sequence in B. licheniformis I5 xylanase is the predicted signal peptide. The amino acid residues identical in all xylanases are black shaded
Comparison between the deduced amino acid sequence of family 11 xylanase of B. licheniformis strain I5 with those of other family 11 xylanase of bacterial origin was shown in Fig. 2. The homology analysis result was as follows: ninety one percent (91%) identity with Bacillus subtilis xylanase, 71% with that of B. halodurans, 72% with that of B. firmus, 90% with that of B.circulans, 71% with that of Thermobacillus xylanilyticus, and 63% with that of Thermobifida fusca. The genes of family 11 endoxylanases were relatively conserved among diverse bacterial resources, as we found when we were designing the primers. BLAST result showed these xylanases genes of family 11 were very highly conserved both at 5′ and 3′ end of the gene among Bacillus species. It was also shown by the alignment of deduced amino acid of our isolated gene with other xylanases. They have very high similarity at N-terminal (or at signal peptide region) and C-terminal as well as at the internal amino acid sequence (Fig. 2).
The results of this study showed that the family 11 endoxylanase might be a ubiquitous enzyme and widely distributed among the microbes inhabited the soil environment such as hot springs. Since the homology of this family of xylanases was high to other xylanases from Bacillus species, it was predicted that the ubiquity was at least among Bacillus species. The ubiquity of lipase A coding gene among Bacillus species living in soil was described by Ruiz et al. (Ruiz et al. 2003). Thus, the same phenomenon might be found in family 11 xylanase of Bacillus species. Therefore, using the specific primers designed in this work would increase the probability of successful isolation of xyn11. Thereby, it will facilitate the PCR-cloning and overexpressing of xyn11 from different species of Bacillus and might accelerate the process in achieving large scale of enzyme production needed for applications.
Comparing the recombinant xylanases of Indonesian B lichenifromis with that of B. halodurans and B firmus, the Indonesian B licheniformis recombinant xylanase has longer nucleotide sequence. Another difference is the internal amino acid (Fig. 2), that our B. licheniformis xylanase does not have cysteine in the peptide chain (Martinez et al. 2005; Chang et al. 2004).
After analysis the activity of cytoplasmic, extratracellular (supernatant), and periplasmic fraction, we found that the enzyme activity was detected mainly in cytoplasmic or intracellular fraction (94.8%). No significant activity detected in extracellular fraction (5% of total activity). Whereas, only 0.02% of total activity detected in periplasmic fraction after osmotic shock. Hence, it is suggested that the existence of the predicted signal peptide of our recombinant xylanase might not be recognized by E. coli. This result was different to that of xylanase from B. subtilis, which was reported to be distributed in intracellular, periplasmic, and extra cellular fraction at significant value (Huang et al. 2005).
The effect of temperature on the activity and thermostability of the recombinant xylanase were observed. The recombinant xylanase has optimum activity at 50°C and pH 7 with the specific activity of 48.4 U/mg (Fig. 3a and b). The activity of recombinant xylanase was higher than that of native crude xylanase which was only 2 U/mg. This result showed that by isolating and expressing the xylanase gene in E. coli, it was possible to increase the productivity of the xylanase, finally it will facilitate the application of the enzyme. The thermostability of the recombinant xylanase was performed by preincubation of crude enzyme extract without substrate at 40, 50, and 55°C for designated time periods and then the enzyme activity was assayed at pH 7, 50°C for 5 min. The enzyme still retained more than 90% of its activity after 30 min incubation at 40°C; however, the residual activity greatly decreased at 50 and 55°C. The residual activity after incubating the enzyme for 10 min at 50°C was 60% but after incubating the enzyme for the same length of time at 55°C, the residual enzyme activity was hardly detectable (Fig. 3c).

Properties of crude extract of recombinant xylanase of B. licheniformis strain I5. (a) Effect of temperature on the activity of recombinant xylanase. For this temperature profile, enzymatic activity was measured with 10 μl crude enzyme extract in 50 mM sodium phosphate buffer (pH 7.0) at different temperatures for 5 min. (b) Effect of pH on the activity of recombinant xylanase in different buffer. The reactions pHs were adjusted to 5–11 with the following buffers: 50 mM Citrate buffer (pH 5, 6) (•), 50 mM Phosphate buffer (pH 6–8) (■), 50 mM Tris-HCl buffer (pH 8–9) (▲), and 50 mM Tris-glycine buffer (pH 10–11) (♦). (c) Thermostability of recombinant xylanase. Thermostability was determined by preincubating 10 μl crude enzyme extract without the substrate at 40°C (♦), 50°C (■), and 55°C (▲) for designated time periods and then assaying the activity in pH 7.0 of 50 mM sodium posphate buffer at 50°C for 5 min as described in Materials and methods. The experiments were conducted in duplicates. Values are the average of two replicates; separate values of the two replicates do not differ more than 10%
The enzyme properties that related to its thermostability and effect of temperature on the activity were nearly similar to that of B. subtilis xylanase. The optimal temperature of B. subtilis xylanase was also at 50°C and it was moderately thermostable at 50°C. However, the optimal pH of B. subtilis xylanase and that of the Indonesian B. licheniformis xylanase were different (Huang et al. 2005).
Actually, the focus of our work was not only to isolate family 11 endoxylanase gene, but also family 10 endoxylanase. Unfortunately, using both degenerated and specific primer PCR-cloning approach we could not obtain the family 10 xylanase gene. Some studies on family 10 endoxylanase (Sunna and Bergquist 2003; Sunna et al. 2000; Ali et al. 2004) mentioned that the enzymes were more thermostable than that of family 11. The molecular mass of the family 10 enzymes were mostly larger than that of the family 11 and some of the family 10 have cellulase activity. Since the family 11 endoxylanases do not have cellulase activity, moreover, they have smaller molecular mass and more compact size than family 10 endoxylanases, therefore they can penetrate easily into the cellulose fiber. Hence, this kind of xylanases is very suitable for many applications (Torronen and Rouvinen 1997; Oakley et al. 2003).
The recombinant xylanase described here showed a good activity at pH 6–8 and a good stability at 40°C, and it also has no damage in cellulose fiber. Therefore, it is expected to be a good candidate for ecologically friendly catalyst in the conversion of agricultural wastes such as water hyacinth stalks or rice straw into more valuable product. These water hyacinth stalks and rice straw are abundantly available in Indonesia (Wiloso et al. 1995). Actually, this recombinant xylanase showed a good potency as a catalyst in pulp-making for high quality art paper from water hyacinth stalks and rice straw (Muksin 2006).
Using E. coli as a host for recombinant xylanase we could improve the enzyme productivity that might be useful for application. Cloning xylanase gene in E. coli also promises further engineering of the enzyme to obtain better enzymes properties. However, in current work the recombinant xylanase was only observed mainly in the intracellular fraction, application using E. coli as a host might not be economically feasible. Therefore, another alternative such as further subcloning of the xylanase gene in mesophilic Bacillus is in progress, so that the gene could be overexpressed as an extracellular enzyme.
References
Agency for Assessment and Application of Technology (BPPT) (2006) Assessement of industrial enzymes market in Indonesia (Indonesian). PT Marina Cipta Pratama, Jakarta
Ali MK, Rudolph FB, Bennett GN (2004) Thermostable xylanase10B from Clostridium acetobutylicum ATCC824. J Ind Microbiol Biotechnol 31:229–234
Bailey MJ, Biely P, Poutanen K (1992) Interlaboratory testing for assay of xylanase activity. J Biochtenol 23:257–270
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Center for Indonesian Geological Environment, Department of Energy and Mineral Resources (Indonesian) (2007) http://www.dgtl.esdm.go.id of subordinate document. Cited 20 July 2007
Chang P, Tsai WS, Tsai CL, Tseng MJ (2004) Cloning and characterization of two thermostable xylanase from an alkaliphilic Bacillus firmus. Biochem Biophys Res Commun 319:1017–1025
Holt JG et al (eds) (1994) Bergey’s manual of determinative bacteriology. Williams & Wilkins, Baltimore, Maryland, USA
Henrissat BA (1991) Classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Huang J, Wang G, Xiao L (2005) Cloning, sequencing, and expression of the xylanase gene from a Bacillus subtilis strain B10 in Escherichia coli. Bioresour Technol 97:802–808
Indonesia Oceanic Cetacean Program Homepage (2007) http://www.apexenvironmental.com/AreaDescription.html. Cited 30 July 2007
Kulkarni NA, Shendye A, Rao M (1999) Molecular and biotechnological aspect of xylanase. FEMS Microbiol Rev 23:411–456
Mahyudin AR, Sita, Mangunwardoyo W (2007) Solid state fermentation as one alternative for xylanase production by thermophilic B. licheniformis I5 with the Banana peels as a substrate. Paper presented at National Meeting of Indonesia Society for Microbiology, Banjarmasin, 30 August–1 September 2007
Martinez MA, Delgado OD, Baigori MD, Sineriz (2005) Sequence analysis, cloning and over-expression of an endoxylanase from the alkalophilic Bacillus halodurans. Biotechnol Lett 27:545–550
Miller GL (1959) Use of dinitrosalycylic acid as reagent for the determination of reducing sugars. Anal Chem 31:208–218
Muksin (2006) Pengolahan serat alami menggunakan system enzim mikrobiologi sebagai media ekspresi seni dua dimensi (Making art paper from natural fiber using microbial enzymes). Laporan Penelitian Program Riset ITB, Dept Seni Rupa, Fakultas Seni Rupa dan Disain, ITB
Oakley AJ, Heinrich T, Thompson CA, Wilce MC (2003) Characterization of a family 11 xylanase from Bacillus subtillis B230 used for paper bleaching. Acta Crystallogr D Biol Crystallogr 59:627–636
Report on Biodiversity and Tropical Forests in Indonesia (2004) http://www.irgltd.com/Resources/Publications of subordinate document. Cited 30 July 2007
Ruiz C, Pastor JFI, Diaz P (2003) Isolation and characterization of Bacillus sp. BP-6 LipA, a ubiquitous lipase among bacillus species. Lett Appl Microbiol 37:354–359
Sambrook J, Fritsch EF, Maniatis T (2001) Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Signal Peptide Prediction Server (2006) http://www.cbs.dtu.dk/services/SignalP/. Cited 10 September 2006
Subramaniyan S, Prema P (2002) Biotechnology of microbial xylanases: enzymology, molecular biology, and application. Crit Rev Biotechnol 22:33–64
Sunna A, Bergquist PL (2003) A gene encoding a novel extremely thermostable 1,4-beta-xylanase isolated directly from an environmental DNA sample. Extremophiles 7:63–70
Sunna A, Gibbs MD, Bergquist PL (2000) A novel thermostable multidomain 1, 4-ß-xylanase from ‘Caldibacillus cellulovorans’ and effect of its xylan-binding domain on enzyme activity. Microbiol 146:2947–2955
Turner P, Mamo G, Karlsson EN (2007) Potential and utilization of thermophile and thermostable enzymes in biorefining. Microb Cell Fact. doi: 10.1186/1475-2859-6-9
Torronen A, Rouvinen J (1997) Structural and functional properties of low molecular weight endo-1, 4-beta-xylanases. J Biotechnol 57:137–49
Wiloso EI, Basuki T, Aiman S (1995) Utilization of agricultural wastes for biogas production in Indonesia. In: Ishizuka et al (eds) Proceedings of the UNESCO—University of Tsukuba international seminar on traditional technology for environmental conservation and sustainable development in the Asian–Pacific region, University of Tsukuba, Japan, 11–14 December, 1995
Acknowledgments
The authors thank to Prof Meinhardt, University of Muenster, for the valuable discussion and Maria Ulfah for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Helianti, I., Nurhayati, N. & Wahyuntari, B. Cloning, sequencing, and expression of a β-1,4-endoxylanase gene from Indonesian Bacillus licheniformis strain I5 in Escherichia coli . World J Microbiol Biotechnol 24, 1273–1279 (2008). https://doi.org/10.1007/s11274-007-9601-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-007-9601-6