
Abstract
In this work, an analytical method for the determination of the 16 polycyclic aromatic hydrocarbons (PAHs), classified as priority pollutants by the US Environmental Protection Agency (EPA), on bitter orange leaves has been optimised and validated. The method has been applied to the evaluation of the applicability of leaves of bitter orange tree as a bioindicator of urban atmospheric pollution by these contaminants. Leaves of bitter orange trees were collected from 13 sampling points in Seville city (South of Spain). Sampling points were located in high-density traffic streets (n = 5), in low-density traffic streets (n = 5) and in urban parks (n = 3). Fourteen of the 16 PAHs monitored were detected in bitter orange leaves. The highest mean concentrations corresponded to BaA, Phen, Pyr and Flt. The concentrations in high-density traffic streets were similar to those in low-density traffic streets. Lower concentrations were found in leaves from parks. PAH diagnostic ratios were applied to identify and to assess pollution emission sources. Diagnostic ratios obtained were consistent with traffic emissions as the main source of PAH to urban air. Based on the obtained results, leaves from bitter orange trees appears to be a promising inexpensive passive sampler suitable for extensive sampling in time and space that can be applied to evaluate risk assessment of urban population to PAH air pollution.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) are persistent organic pollutants formed in incomplete combustion on natural (forest fires, volcanic eruptions, etc.) and anthropogenic (aluminium, iron and steel production in plants and foundries; waste incineration; mining; oil refining; cigarette smoke; burning of wood for heating and motor vehicle emissions) processes (Kiss et al. 2001; Krauss et al. 2005; Orecchio et al. 2016; Xu et al. 2006). PAHs have been identified as carcinogenic, mutagenic and teratogenic (Straif et al. 2006) and show a leading role on several human health risks, especially oncogenic risk. They are present in urban air from most of the big cities around the world (Lee et al. 2004; Menichini et al. 2007).
The studies about PAH air pollution have been usually carried out by their analysis in air or in particulate matter. These studies provide useful information about their distribution in the inhalable, thoracic and breathable fractions. However, the applied analytical methods have some limitations, mainly related to the complexity of the sampling process (diffuse sources, meteorology, etc.) and the low concentrations of these pollutants. These difficulties can be overcome with the use of living organisms as bioindicators and bioaccumulators (Käffer et al. 2012; Ukpebor et al. 2010). The use of tree leaves as bioindicators of atmospheric pollution constitute an excellent and cost-effective way to detect and assess environmental pollution, especially in developing countries and remote regions. Moreover, evergreen species can be used to evaluate pollutant accumulation through long-term exposure (Murakami et al. 2012; Orecchio and Culotta 2015). It is an effective tool to detect potential health and environmental risks to the vicinity of pollutant emission sources (Rodriguez et al. 2012).
In 2004, the European Parliament published the Directive 2004/107/EC in which the use of bioindicators for assessing the PAH impact on ecosystems was recommended. Considering specifically the bioaccumulation of PAHs in tree leaves, it is possible to consider the vegetation as a large collector of hydrocarbons. The absorption of pollutants can occur from air (atmospheric deposition on plants, especially on leaves) and from soil (through xylem transport). This last bioaccumulation way is less significant than the first one in the case of PAHs, because their lipophilic character makes difficult their transport through the xylem (Amand et al. 2009).
In the last years, several analytical methods have been described in the literature for the determination of PAHs in leaves from several tree species. Because of the complexity of the matrix, accurate and sensitive analytical methods are necessary for the determination of PAHs in leaves. Gas chromatography coupled to mass spectrometry (GC-MS) (Käffer et al. 2012; Krauss et al. 2005; Lehndorff and Schwark 2004; Orecchio 2007; Prajapati and Tripathi 2008; Sun et al. 2010; Tomashuk et al. 2012) and high-performance liquid chromatography with diode array (HPLC-DAD) and/or fluorescence detectors (HPLC-Fl) (Augusto et al. 2010; Rodriguez et al. 2012) are the most used techniques for this purpose. The extraction techniques most commonly applied to the extraction of PAHs are ultrasonic-assisted extraction (Käffer et al. 2012; Murakami et al. 2012; Orecchio 2007; Prajapati and Tripathi 2008; Rodriguez et al. 2012; Yin et al. 2011), accelerated solvent extraction (Amand et al. 2009; Lehndorff and Schwark 2004) or Soxhlet extraction (Augusto et al. 2010; Sun et al. 2010). Most of these methods use large volumes of extraction solvents (higher than 40 mL) (Augusto et al. 2010; Käffer et al. 2012; Murakami et al. 2012; Orecchio 2007; Prajapati and Tripathi 2008) and chlorinated solvents such as dichloromethane or mixtures of dichloromethane and hexane or acetone (Amand et al. 2009; Prajapati and Tripathi 2008; Käffer et al. 2012; Lehndorff and Schwark 2004; Murakami et al. 2012; Orecchio 2007; Sun et al. 2010; Tomashuk et al. 2012). These analytical methods have been applied for biomonitoring of PAHs in trees such as pine (Augusto et al. 2010; Lehndorff and Schwark 2004; Rodriguez et al. 2012; Sun et al. 2010; Wang et al. 2009), gingko (Murakami et al. 2012; Yin et al. 2011), eucalyptus, populous (Rodriguez et al. 2012) and quercus (Orecchio 2007). Nevertheless, to the best of our knowledge, no study has been done using leaves from bitter orange tree (Citrus aurantium) for biomonitoring PAH air pollution. The bitter orange tree is an evergreen small tree (height from less than 3 to 9 m) that it is grown throughout Asia, North Africa, the Mediterranean region and South America. This tree species can be found in parks, in main streets and in pedestrian areas from urban and sub-urban areas. For this reason, this species can be particularly suitable as a bioindicator of PAH air pollution and useful for extensive sampling in time and space.
The aim of this work was to evaluate the applicability of bitter orange leaves as bioindicators of PAHs in urban air. To achieve this goal, an analytical method, based on ultrasonic-assisted extraction and determination by high-performance liquid chromatography (HPLC) with diode array and fluorescence detectors, has been developed. The method was applied to obtain information about PAH concentration levels in urban air and their relation to traffic density.
2 Experimental
2.1 Reagents and Materials
HPLC-grade acetone, acetonitrile, dichloromethane, hexane, methanol and water were purchased from Romil Ltd. (Barcelona, Spain). Analytical-grade sodium sulphate was obtained from Scharlab (Barcelona, Spain). A PAH standard mixture (EPA 8270 PAH mix 3) (naphthalene (Naph, 1000 mg/L), acenaphthylene (Acy, 2000 mg/L), acenaphthene (Ace, 1000 mg/L), fluorene (Fluo, 100 mg/L), phenanthrene (Phen, 100 mg/L), anthracene (Ant, 100 mg/L), fluoranthene (Flt, 100 mg/L), pyrene (Pyr, 100 mg/L), benzo(a)anthracene (BaA, 100 mg/L), chrysene (Chry, 100 mg/L), benzo(b)fluoranthene (BbF, 100 mg/L), benzo(k)fluoranthene (BkF, 100 mg/L), benzo(a)pyrene (BaP, 100 mg/L), dibenzo(a,h)anthracene (DahA, 200 mg/L), benzo(g,h,i)perylene (BghiP, 100 mg/L) and indeno(1,2,3-cd)pyrene (IcdP, 100 mg/L) in methylene chloride/methanol (1:1; v/v)) was purchased from Sigma-Aldrich (Steinheim, USA). BAKERBOND octadecyl C18 sorbent (40 μm particle size) was provided by J.T. Baker (Deventer, The Netherlands). Nylon syringe filters (0.45 μm pore size) were supplied by Scharlau (Barcelona, Spain).
PAH stock solutions were prepared by dilution of EPA 8270 PAH mix 3 in acetonitrile. Stock solutions were stored at 4 °C. Working solutions were prepared by diluting the stock standard solutions in methanol.
2.2 Studied Area and Sampling Points
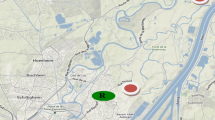
Bitter orange leaves were sampled from 13 sites in Seville city (South of Spain) covering a superficial area of about 28 km2. Sampling points were selected taking into account traffic data from 2006 to 2011 from the site of the municipality of Seville (http://www.seville.org). Sampling points were located in five high-density traffic streets, in five low-density traffic streets, in two urban parks and in one extra-urban park. The five high-density streets selected, in increasing order of traffic density, were: Puente Cristo de la Expiración (HT1), Paseo Colón (HT2), Torneo (HT3), Delicias-Palmera (HT4) and Luis Montoto (HT5). The five low-density streets selected, in increasing order of traffic density, were: El Greco (LT1), Ronda de Triana (LT2), Buhaira (LT3), Ciudad Jardín (LT4) and Asunción (LT5). The urban parks selected were Los Príncipes (P1) and María Luisa (P2). The extra-urban park selected was Alamillo (P3). Figure 1 shows the studied area and streets locations. For each sampling site, 25 % of the bitter orange trees were sampled. Trees with anomalies, gummosis or putrefaction of the neck of the root, infections caused by viruses or presence of parasites were discarded.

Studied area and street locations
Five leaves, one from each cardinal point and one from the centre of the tree, were collected from each sampled tree. Then, composite sample, containing all the leaves from each sampling site, were obtained. Composite samples were transported to the laboratory on plastic bags (25 × 30 cm) with airtight seal. The samples were weighed, washed with two portions of deionised water to remove deposed PAH, dried, cut, lyophilised (0.01 mbar vacuum after being frozen at −18 °C for 24 h), pulverised and sieved (<1 mm).
2.3 Sample Treatment
Leaves (1.0 g) were mixed with 0.5 g of sodium sulphate and extracted consecutively with three portions of 3 mL of acetone. In each extraction step, sample was vigorously shaken in a vortex-mixer during 30 s, sonicated for 10 min and centrifuged at 4000 rpm for 20 min. The supernatants obtained from each extraction step were combined and cleaned-up by dispersive solid-phase extraction (dSPE). The extract was transferred to a 10-mL glass conical tube containing 500 mg of C18 sorbent. The mixture was hand-shaken for 2 min and centrifuged for 10 min at 4000 rpm. The organic phase was transferred to a glass vial and evaporated to dryness by a gentle nitrogen stream, dissolved in 0.3 mL of methanol and filtered through a 0.45-μm filter.
2.4 HPLC-DAD-Fl
Chromatographic analyses were performed using an HPLC 1200 Series instrument (Agilent, USA) equipped with a G1321A binary pump, a G1329A automatic injector, a G1379B degasser, a G1316A thermostated column compartment, a G1315D ultraviolet diode array detector (DAD) and a G1321A fluorescence scanning detector (Fl) connected on line. Separations were carried out using a LiChrospher® PAH (250 mm × 3 mm i.d., 5 μm) column (Merck, Darmstadt, Germany). Cartridge column was protected by a LiChrospher® 100 RP-18 (4 mm × 4 mm i.d., 5 μm) guard column (Merck, Darmstadt, Germany). The system was controlled by an interface module and a personal computer. Chromatograms were processed by ChemStation software (Agilent).
Analytes were separated by gradient elution with acetonitrile (A) and HPLC-grade water (B) at a flow-rate of 1 mL/min with the column thermostated at 30 °C. The elution programme was: 0–3 min, isocratic 60 % of solvent A; 3–15 min, linear gradient from 60 to 100 % of solvent A and 15–20 min, isocratic 100 % of solvent A. Ultraviolet (UV) chromatogram was monitored using the UV signal at 254 nm. Quantification was carried out using fluorescence signal at the optimal excitation and emission wavelengths (Table 1), except for Acy. UV signal was used for Acy determination because it is not fluorescent. The calibration curves were constructed in the concentration range expected of each PAH. Calibration standards were prepared by dilution of the stock standard solutions in methanol. Calibration curves were generated by linear regression of peak areas of standard solutions against their respective concentrations.
2.5 Method Validation
The analytical method was validated by the determination of recovery, precision (expressed as repeatability in terms of relative standard deviation (RSD)), method working range, linearity and method detection (MDL) and quantification limits (MQL). Recoveries were evaluated by comparison between the peak area of the analyte in quintuplicate spiked samples (A) and the area of the analyte in a standard solution at the same concentration level (B): R (%) = (A/B) × 100. Blank samples (non-spiked samples) were measured to apply blank corrections to signals obtained in spiked samples. Precision of the method was calculated as the RSD of the concentrations determined in spiked samples measured in triplicate. Instrument working range was evaluated by duplicate injection of seven standard solutions at concentrations ranging from instrumental quantification limit (IQL) to 500 μg/L. Method working range was calculated from instrumental working range taking into account the concentration factor achieved in sample treatment and the recovery of each PAH. Calibration curves were constructed by linear regression of the peak areas of each analyte against their respective concentrations. The concentrations of the target compounds in the studied samples were calculated from the concentrations determined in sample extracts taking into account the concentration factor of sample treatment and the recovery of the target compound. Each analysis was carried out in triplicate in order to avoid false positives. Instrumental detection limit (IDL) and IQL were estimated as the concentrations of each analyte corresponding to a signal-to-noise ratio of 3:1 and 10:1, respectively. ILD and ILQ were determined by quintuplicate injections of standard solutions at low concentration levels. MDL and MQL were calculated from ILD and ILQ applying the concentration factor and recoveries achieved in sample treatment. Procedural blanks were also processed for signal correction.
2.6 Data Analysis
Data analysis was carried out using graphical and statistical tools. Statistical techniques were used to establish relations between concentration levels of PAHs from different sampling sites. Correlation and factorial analysis were performed using Statistical 6.0 software for Windows.
3 Results and Discussion
3.1 Sample Treatment Optimization
Acetone, acetonitrile, hexane and mixture acetone/hexane (1:1, v/v) were tested as extraction solvents. Portions of 1.0 g dm of bitter orange leaves were spiked in triplicate with PAHs at a concentration level of 100 μg/kg dm. Spiked and blank samples were successively extracted with three aliquots of 3 mL of the tested solvent by sonication for 10 min. Recoveries obtained are shown in Fig. 2. Poor recoveries were achieved when hexane and acetone/hexane (1:1, v/v) mixture (mean recoveries 25 and 61 %, respectively) were used as extraction solvents. The highest recoveries were obtained using acetonitrile (ranged 14–114 %) and acetone (55–102 %). However, DahA, BghiP and IcdP were poorly extracted using acetonitrile. Precision (expressed as RSD (%)) obtained with acetonitrile (mean precision 28 %) was poorer than that obtained with acetone (mean precision 11 %). Based on these results, acetone was chosen as extraction solvent.

Influence of extraction solvent (n = 3) and clean-up method (n = 3) on extraction recoveries
Two procedures were tested for extract clean-up: solid-phase extraction (SPE) and dSPE. SPE clean-up was carried out with Oasis HLB cartridges (packed with 60 mg of Oasis HLB sorbent, purchased from Waters (Milford, MA, USA)). SPE cartridges were conditioned with 3 mL of acetone and 3 mL methanol. Then, the extract obtained after sonication extraction was percolated at a flow-rate of about 3 mL/min through the cartridge, evaporated to dryness by a gentle nitrogen stream, dissolved in 0.5 mL of methanol and filtered through a 0.45-μm filter. dSPE was carried out with C18 sorbent as described in Section 2.3. dSPE was selected as clean-up procedure due to the higher recoveries (Fig. 2), quickness and low-cost.
3.2 Method Performance
Table 1 shows recovery, precision (expressed as repeatability in terms of RSD), method working range, linearity and MDL and MQL.
Recovery and precision were evaluated by quintuplicate analysis of samples spiked with the studied compounds at 100 μg/kg dm. The recoveries achieved with the proposed method were higher than 74 % for all studied PAHs, except for Naph (55 %) and IcdP (66 %). Precision, measured as RSD, was lower than 5 % for 13 of the studied PAH. Only two of the studied compounds (Naph and Acy) had precisions higher than 10 %. The recoveries and precision were similar to those reported by other authors using time consuming extraction methods (Augusto et al. 2010; Käffer et al. 2012; Orecchio 2007) and high volumes of toxic solvents such as dichloromethane (Murakami et al. 2012; Orecchio 2007; Prajapati and Tripathi 2008). Calibration curves were constructed by the duplicate injection of standard solutions at concentrations in the range from IQL to 500 μg/L. The highest calibration points, covering the purpose of the method, were in the range from 147 (Phen) to 273 μg/kg dm (Naph). Calibration curves were linear over the concentration range studied with correlation coefficients greater than 0.99 for all of the studied PAHs. MDL were ranged from 0.03 μg/kg dm (BkF) to 0.62 μg/kg dm (Acy) and MQL were ranged from 0.09 μg/kg dm (BkF) to 2.29 μg/kg dm (IcdP). These MDL and MQL were similar to those previously reported by several authors, in spite of the low sample amount required by the proposed method (1 g) in comparison to sample amounts in other methods that are higher than 5 g (Amand et al. 2009; Murakami et al. 2012; Orecchio 2007; Prajapati and Tripathi 2008; Sun et al. 2010).
3.3 PAH Concentration in Bitter Orange Leaves
All studied compounds, except Acy, BaP and IcdP, were found in the analysed leaves (Table 2). PAH concentrations in high-density traffic streets (mean concentration 205.7 μg/kg dm, expressed as the sum of the analysed PAH) were similar to those in low-density traffic streets (202.5 μg/kg dm, expressed as the sum of the analysed PAH). This fact could be explained by the different geometry of the studied streets. Most of the low-density traffic streets studied were narrower and with tall buildings, and therefore worse ventilated, than the studied high-density traffic streets. This fact could cause pollutant accumulation in low-density traffic streets. The lowest concentrations were measured in parks (mean concentration 121.0 μg/kg dm). The highest mean concentrations corresponded to BaA (96.3 μg/kg dm), Phen (28.9 μg/kg dm), Pyr (26.3 μg/kg dm) and Flt (12.7 μg/kg dm), which, additionally, were detected in all sampling sites. The other PAH detected in all sampling sites were Fluo (mean concentration 2.6 μg/kg dm), Ant (mean concentration 2.0 μg/kg dm), Ace (mean concentration 0.8 μg/kg dm) and BkF (mean concentration 0.6 μg/kg dm). The results reported in this work were comparable with data obtained from other bioindicators such as pine needles (Wang et al. 2009; Lehndorff and Schwark 2004), gingko leaves (Murakami et al. 2012; Yin et al. 2011) and eucalyptus (Rodriguez et al. 2012). For instance, Phen was the predominant PAH followed by Pyr and Flt in pine needles (Lehndorff and Schwark 2004). Other studies reported BaA (Tomashuk et al. 2012) and Fluo (Rodriguez et al. 2012) as the predominant PAHs. The higher concentrations of three- and four-ring PAHs can be explained by an easier contact with leaves than heaviest PAHs. Three- and four-ring PAH mainly exists in the gas phase and can be easily sequestered by leaves than five- and six-ring PAHs which are mainly associated with particles and can easily deposit into soils (Wang et al. 2009; Tomashuk et al. 2012).
3.4 Sources of Bitter Orange Leave Exposition to PAHs
PAH sources were estimated from the ratio of low and high molecular weight PAH (Barreca et al. 2014), distribution indexes (Barreca et al. 2014) and PAH ratios (Barreca et al. 2014; Tobiszewski and Namiesnik 2012; Yunker et al. 2002). The ratio of low and high molecular weight PAH (Acy + Ace + Fl + Phen + Ant)/(BaA + Cry + BbF + BkF + BaP + InP + DBA + BgP) calculated was 0.26, indicating pyrogenic as the main PAH source (Barreca et al. 2014). Distribution indexes are based on the thermodynamic stability of the compounds and are related to concentration ratios of some single compounds (Barreca et al. 2014). They can be useful for identifying a given emission source. The total index, calculated according to Barreca et al. (2014), (total index = Ant/(Ant + Phen)/0.1 + Flt/(Flt + Pyr)/0.4 + BaA/(BaA + Chry)/0.2 + IcdP/(IcdP + BghiP)/0.5) was 6.36, which indicates combustion source. Fluo/(Fluo + Pyr), Flt/(Flt + Pyr) and BaA/(BaA + Chry) ratios (Barreca et al. 2014; Tobiszewski and Namiesnik 2012; Yunker et al. 2002) were also calculated. PAH ratios have even been used to distinguish between diesel and gasoline combustion emissions (Ravindra et al. 2008). Fluo/(Fluo + Pyr) ratios lower than 0.5 are considered indicators of gasoline emission whilst values higher than 0.5 indicate diesel emission (de la Torre-Roche et al. 2009). Flt/(Flt + Pyr) ratio values lower than 0.4 indicates petroleum input, ratios between 0.4 and 0.5 can be associated to liquid fossil fuel (vehicle and crude oil) combustion (Pies et al. 2008; Ravindra et al. 2008) and ratios higher than 0.5 can indicate grass, wood or coal combustion (de la Torre-Roche et al. 2009; Katsoyiannis et al. 2007). BaA/(BaA + Chry) ratio values lower than 0.2 indicate vehicular emission (Pies et al. 2008), values between 0.2 and 0.35 indicate coal combustion (Tobiszewski and Namiesnik 2012) and values higher than 0.35 indicate fuel combustion (Yunker et al. 2002). Fluo/(Fluo + Pyr) and BaA/(BaA + Chry) ratio values obtained were 0.09 and 0.98, which could indicate diesel emission and combustion, respectively. Flt/(Flt + Pyr) values were close to 0.4 (mean 0.37) which can be associated with liquid fossil fuel (vehicle and crude oil) combustion.
These results are consistent with the fact that vehicular emission is the main source of PAHs in urban air pollution in the area sampled in this study. Nevertheless, the diagnostic ratios should be used with caution in studies of bioindicators, firstly, because it is often difficult to discriminate between sources (Ravindra et al. 2008) and, secondly, because the reactivity of some PAH in the atmosphere together with their distribution in leaves can change diagnostic ratio values (Ravindra et al. 2008) from ratio values on soil (Gutierrez-Daban et al. 2005; Wang et al. 2009).
In order to establish a possible relation between traffic density and PAH concentration, a statistic data treatment, based on correlation analysis and factorial analysis, was carried out. Correlation analysis was carried out considering sampling points as variables and PAH concentrations and traffic density as cases. Results of the correlation analysis are shown in Table 3. High correlations (>0.97) were found between sampling points HT1–HT5 and LT1–LT3 whilst low correlations were found between HT1–HT5 and LT4–LT5 and parks (P1–P3). Moreover, high correlations were found between P1 and LT4 (0.91) and between P1–P2 and LT5.
Factorial analysis was carried out considering sampling points as variables and PAH concentrations as cases. Two factors with eigenvalues >1 (factor 1 = 7.98; factor 2 = 3.06), accounting for 85 % of the total variability, were identified (Table 4). The first component reflects a close correlation between sampling points with the highest traffic density (HT1–HT5 and LT1–LT3) whereas the second factor reflects a close correlation between sampling points with the lowest traffic density (LT4, LT5 and parks). In Fig. 3, the correlations between the investigated variables and the two factors are represented. Variables were mainly grouped into two groups, one group formed by HT sampling points and LT1–L3, and another group formed by LT5 and P2 and P3. A low contribution of LT4 and P1 to these factors was observed.

Plot of the variables on the plane factor 1 versus factor 2
These relations could be explained by the influence of traffic on PAH concentrations in streets with the highest traffic density and by the similar low PAH concentration in parks P2 and P3 and LT5 (pedestrian street). The low contribution of P1 and LT4 to the obtained factors could be due to an intermediate level of contamination by PAHs in these sampling points.
4 Conclusions
An analytical method has been developed for the determination of PAH in bitter orange leaves. Sample treatment is based on ultrasonic solvent extraction and clean-up by dSPE. Analytical determination was carried out by HPLC-DAD-Fl. The method was developed for the determination of the 16 PAHs considered priority pollutants by the Environmental Protection Agency. The method was successfully applied for the determination of PAHs in bitter orange leaves from trees located in high-density streets, low-density streets and parks. PAH concentration effect was observed in low-density traffic streets when compared with high-density traffic streets. Higher concentrations were observed for lightest PAHs; it is three- and four-ring PAH, than for five- and six-ring PAH. Concentrations obtained were consistent with results reported in other bioindicators such as pine needles. Diagnostic ratios and statistical analysis were consistent with vehicular emission as the main pollution emission source of PAH in the urban atmosphere. This study marks a starting point for the use of bitter orange leaves as an alternative to the high-cost instrumental for extensive sampling, in time and/or in space, of PAH in urban air. More studies are needed to extend the applicability of bitter orange leaves for biomonitoring of other pollutants in the urban atmosphere.
References
Amand, A. D., Mayer, P. M., & Blais, J. M. (2009). Modeling PAH uptake by vegetation from the air using field measurements. Atmospheric Environment, 43, 4283–4288.
Augusto, S., Maguas, C., Matos, J., Pereira, M. J., & Branquinho, C. (2010). Lichens as an integrating tool for monitoring PAH atmospheric deposition: a comparison with soil, air and pine needles. Environmental Pollution, 158, 483–489.
Barreca, S., Bastone, S., Caponetti, E., Martino, D. F. C., & Orecchio, S. (2014). Determination of selected polyaromatic hydrocarbons by gas chromatography–mass spectrometry for the analysis of wood to establish the cause of sinking of an old vessel (Scauri wreck) by fire. Microchemical Journal, 117, 116–121.
de la Torre-Roche, J. R., Lee, W.-Y., & Campos-Díaz, S. I. (2009). Soil-borne polycyclic aromatic hydrocarbons in El Paso, Texas: analysis of a potential problem in the United States/Mexico border region. Journal of Hazardous Materials, 163, 946–958.
Gutierrez-Daban, A., Fernandez-Espinosa, A. J., Ternero-Rodrıguez, M., & Fernandez-Alvarez, F. (2005). Particle-size distribution of polycyclic aromatic hydrocarbons in urban air in southern Spain. Analytical and Bioanalytical Chemistry, 381, 721–736.
Käffer, M. I., Lemos, A. T., Apel, M. A., Rocha, J. V., Martins, S. M. A., & Vargas, V. M. F. (2012). Use of bioindicators to evaluate air quality and genotoxic compounds in an urban environment in Southern Brazil. Environmental Pollution, 163, 24–31.
Katsoyiannis, A., Terzi, E., & Cai, Q.-Y. (2007). On the use of PAH molecular diagnostic ratios in sewage sludge for the understanding of the PAH sources. Is this use appropriate? Chemosphere, 69, 1337–1339.
Kiss, G., Varga-Puchony, Z., Tolnai, B., Varga, B., Gelencsér, A., Krivàcsy, Z., & Hlavay, J. (2001). The seasonal changes in the concentration of polycyclic aromatic hydrocarbons in precipitation and aerosol near Lake Balaton, Hungary. Environmental Pollution, 114, 55–61.
Krauss, M., Wilcke, W., Martius, M., Bandeire, A. G., Garcia, M. V. B., & Amelung, W. (2005). Atmospheric versus biological sources of polycyclic aromatic hydrocarbons (PAHs) in a tropical rain forest environment. Environmental Pollution, 135, 143–154.
Lee, J. H., Gigliotti, C. L., Offenberg, J. H., Eisenreich, S. J., & Turpin, B. J. (2004). Sources of polycyclic aromatic hydrocarbons to the Hudson River Airshed. Atmospheric Environment, 38, 5971–5981.
Lehndorff, E., & Schwark, L. (2004). Biomonitoring of air quality in the Cologne Conurbation using pine needles as a passive sampler—Part II: polycyclic aromatic hydrocarbons (PAH). Atmospheric Environment, 38, 3793–3808.
Menichini, E., Iacovella, N., Monfredini, F., & Turrio-Baldassarri, L. (2007). Atmospheric pollution by PAHs, PCDD/Fs and PCBs simultaneously collected at a regional background site in central Italy and at an urban site in Rome. Chemosphere, 69, 422–434.
Murakami, M., Abe, M., Kakumoto, Y., Kawano, H., Fukasawa, H., Saha, M., & Takada, H. (2012). Evaluation of ginkgo as a biomonitor of airborne polycyclic aromatic hydrocarbons. Atmospheric Environment, 54, 9–17.
Orecchio, S. (2007). PAHs associated with the leaves of Quercus ilex L.: extraction, GC–MS analysis, distribution and sources: assessment of air quality in the Palermo (Italy) area. Atmospheric Environment, 41(38), 8669–8680.
Orecchio, S., & Culotta, L. (2015). Assessment of quality of air in Palermo by chemical (ICP-OES) and cytological analyses on leaves of Eucalyptus camaldulensis. Environmental Science and Pollution Research, 22, 1891–1905.
Orecchio, S., Amorello, D., Barreca, S., & Valenti, A. (2016). Wood pellets for home heating can be considered environmentally friendly fuels? Polycyclic aromatic hydrocarbons (PAHs) in their ashes. Microchemical Journal, 124, 267–271.
Pies, C., Hoffmann, B., Petrowsky, J., Yang, Y., Ternes, T. A., & Hofmann, T. (2008). Characterization and source identification of polycyclic aromatic hydrocarbons (PAHs) in river bank soils. Chemosphere, 72, 1594–1601.
Prajapati, S. K., & Tripathi, B. D. (2008). Biomonitoring seasonal variation of urban air polycyclic aromatic hydrocarbons (PAHs) using Ficus benghalensis leaves. Environmental Pollution, 151, 543–548.
Ravindra, K., Wauters, E., & Van Grieken, R. (2008). Variation in particulate PAHs levels and their relation with the transboundary movement of the air masses. Science of the Total Environment, 396, 100–110.
Rodriguez, J. H., Wannaz, E. D., Salazar, M. J., Pignata, M. L., Fangmeier, A., & Franzaring, J. (2012). Accumulation of polycyclic aromatic hydrocarbons and heavy metals in the tree foliage of Eucalyptus rostrata, Pinus radiata and Populus hybridus in the vicinity of a large aluminium smelter in Argentina. Atmospheric Environment, 55, 35–42.
Straif, K., Baan, R., Grosse, Y., Secretan, B., El Ghissassi, F., & Cogliano, V. (2006). Carcinogenicity of household solid fuel combustion and of high-temperature frying. Lancet Oncology, 7, 977–978.
Sun, F., Wen, D., Kuang, Y., Li, J., Li, J., & Zuo, W. (2010). Concentrations of heavy metals and polycyclic aromatic hydrocarbons in needles of Masson pine (Pinus massoniana L.) growing nearby different industrial sources. Journal of Environmental Science, 22, 1006–1013.
Tobiszewski, M., & Namiesnik, J. (2012). PAH diagnostic ratios for the identification of pollution emission sources. Environmental Pollution, 162, 110–119.
Tomashuk, T. A., Truong, T. M., Mantha, M., & McGowin, A. E. (2012). Atmospheric polycyclic aromatic hydrocarbon profiles and sources in pine needles and particulate matter in Dayton, Ohio, USA. Atmospheric Environment, 51, 196–202.
Ukpebor, E. E., Ukpebor, J. E., Aigbokhan, E., Goji, I., Onojeghuo, A. O., & Okonkwo, A. C. (2010). Delonix regia and Casuarina equisetifolia as passive biomonitors and as bioaccumulators of atmospheric trace metals. Journal of Environmental Science, 22, 1073–1079.
Wang, Z., Chen, J., Yang, P., Tian, F., Qiao, X., Bian, H., & Ge, L. (2009). Distribution of PAHs in pine (Pinus thunbergii) needles and soils correlates with their gas-particle partitioning. Environmental Science and Technology, 43, 1336–1341.
Xu, S. S., Liu, W. X., & Tao, S. (2006). Emission of polycyclic aromatic hydrocarbons in China. Environmental Science and Technology, 40, 702–708.
Yin, H., Tan, Q., Chen, Y., Lv, G., & Hou, X. (2011). Polycyclic aromatic hydrocarbons (PAHs) pollution recorded in annual rings of gingko (Gingko biloba L.): determination of PAHs by GC/MS after accelerated solvent extraction. Microchemical Journal, 97, 138–143.
Yunker, M. B., Macdonald, R. W., Vingarzan, R., Mitchell, R. H., Goyette, D., & Sylvestre, S. (2002). PAHs in the Fraser River basin: a critical appraisal of PAH ratios as indicators of PAH source and composition. Organic Geochemistry, 33, 489–515.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fasani, D., Fermo, P., Barroso, P.J. et al. Analytical Method for Biomonitoring of PAH Using Leaves of Bitter Orange Trees (Citrus aurantium): a Case Study in South Spain. Water Air Soil Pollut 227, 360 (2016). https://doi.org/10.1007/s11270-016-3056-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-016-3056-z