
Abstract
Porcine reproductive and respiratory syndrome (PRRS) is an emerging disease that has caused serious economic losses to the swine industry worldwide. In 2011, a nation-wide surveillance program investigated the prevalence of PRRS viruses (PRRSV) in Chinese breeding swine farms, and four European genotype PRRSV (PRRSV-Type 1) were successfully isolated. To explore the genetic diversity of PRRSV-Type 1 in China, these 4 viral strains were subjected to genome sequencing and analysis. The four isolates shared 87.4–90.7 % nucleotide homology with the Lelystad strain (PRRSV-Type 1 stereotype strain). NSP2, ORF3, and ORF4 were the most variable regions and contained discontinuous deletions or insertions when compared to other PRRSV-Type 1 strains. All isolates fell into separate branches of the subtype 1 of PRRSV-Type 1 phylogenetic tree. This analysis of emerging PRRSV-Type 1 strains revealed previously unrecorded genetic diversity. Close attention should be paid to the prevention and control of this evolving virus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is one of the most important swine infectious diseases and causes great economic losses in the swine industry worldwide [1, 2]. PRRS virus (PRRSV) is a member of the Arterivirus genus, family Arteriviridae, order Nidovirales, and is a small enveloped virus with a single-stranded positive-sense RNA genome [3]. The genome of PRRSV is approximately 15 kb and contains at least ten overlapping open reading frames (ORFs), including ORF1a, ORF1b, ORF2a, ORF2b, ORF3, ORF4, ORF5a, ORF5, ORF6, and ORF7 [4–6]. Of these, ORF1a and ORF1b are located at the 5′ terminal of the genome and encode viral nonstructural proteins (Nsp), while ORFs2-7 are located at the 3′ terminal region of the genome, and encode minor structural proteins (GP2, GP3, and GP4) and major structural proteins(GP5a, GP5, M, and N) [2, 4, 7].
PRRSV genome sequences are highly diverse and can be divided into European genotype (PRRSV-Type 1) and North American genotype (PRRSV-Type 2) [8, 9]. PRRSV-Type 1 first emerged in Europe, and the Lelystad virus represents the prototypical Type 1 strain [1]. PRRSV-Type 2 emerged in North America, and the ATCC VR-2332 strain represents the prototype Type 2 virus [10]. Originally, PRRSV-Type 1 viruses were only detected in European swine herds. However, PRRSV-Type 1 has recently been reported in North America and Asian swine herds, and the coexistence of these two genotypes in swine herds represents a huge challenge for PRRSV differential diagnosis, and disease prevention and control [11–16].
In China, the emergence of the PRRSV-Type 1 strains has been previously reported [17], but most PRRSV isolates have been of the PRRSV-Type 2 genotype, and the origin and molecular evolution of PRRSV-Type 1 in China is currently unknown. To gain further insight into the epidemic status and genetic divergence of PRRSV-Type 1 viruses in China, 4 PRRSV-Type 1 viruses were isolated in 2011, and subjected to genomic analysis.
Materials and methods
Ethics statement
The 4136 marginal ear vein blood samples were collected from 80 breeding swine farms in China. Since this study was carried out in private farms, the animals were sampled with the permission of the farm managers. The animal field trial was approved by the Animal Care and Ethics Committee of China Animal Disease Control Center and conventional animal welfare regulations and standards were adhered to.
Virus isolation
The serum samples were screened for both PRRSV genotypes by conventional RT-PCR [12]. Samples that tested positive for PRRSV were screened by a real-time RT-PCR for PRRSV-Type 1 and PRRSV-Type 2 [18]. PRRSV-Type 1 positive serum samples were inoculated on primary porcine alveolar macrophages (PAMs) and Marc-145 cells for virus isolation as previously described [19, 20]. Inoculated cells were maintained at 37 °C in a 5 % CO2 atmosphere and were monitored daily for cytopathic effects (CPE). When CPE were observed in 70 % of the cells, cultures were harvested and the resultant virus stocks were stored at −80 °C.
RNA extraction and genome sequencing
Total RNA was extracted from cell cultures using an RNeasy Mini kit (Qiagen, Germany) according to the manufacturer’s instructions. Whole genomes were sequenced as previously described [17]. Briefly, 16 overlapping fragments covering the whole viral genome were amplified by RT-PCR using the corresponding primers. PCR products were purified with E.Z.N.A.TM Gel Extraction Kit (Omega, USA) and were cloned into pEASY-T1 (Transgen, China). The recombinant plasmids were sequenced with an ABI Automatic DNA Sequencer (Invitrogen, China) and artificially spliced. Gene fragments were sequenced in triplicate and were assembled with ContigExpress in NTI Advance 11Vector [21].
Multiple sequence alignments and phylogenetic analysis
Multiple alignments were performed for each ORF separately using all available PRRSV-Type 1 virus whole genome sequences in GenBank (Suppl. Table). Phylogenetic trees were constructed from aligned nucleotide sequences using the neighbor-joining method, and were subsequently subjected to bootstrap analysis with 1000 replicates to determine the percentage of reliabilities at each internal node of the tree. The tree was produced using the MEGA4.1 program [22].
Results
Four PRRSV-Type 1 viruses were isolated in a national epidemiological survey
To explore the prevalence of PRRSV-Type 1 virus infection in Chinese swine herds, 4136 serum samples were collected from 80 breeding swine farms in different provinces. All samples were subjected to conventional RT-PCR for both genotypes of PRRSV, and samples in which PRRSV was detected underwent real-time RT-PCR for PRRSV-Type 1 and PRRSV-Type 2 differentiation [12, 18]. PRRSV-Type 2 virus was detected in 18/80(22.5 %) breeding swine farms and PRRSV-Type 1 virus was detected in 8/80(10 %) farms in China. The PRRSV-Type 1 virus-positive serum samples were subsequently inoculated on primary PAM and Marc-145 cells. After 3 days, four inoculates exhibited a cytotoxic effect on PAMs but not on Marc-145 cells. Moreover, these isolates were labeled by PRRSV-specific monoclonal antibodies, further confirming the presence of PRRSV, and excluding contamination with other swine viruses (data not show). These 4 PRRSV isolates were designated NVDC-NM1-2011, NVDC-NM2, NVDC-NM3, and NVDC-FJ.
Genomic sequence analysis of 4 PRRSV-Type 1 isolates
The complete genomic sequences of four viral isolates were assembled into contiguous sequences of 15081(NVDC-NM1-2011, GenBank accession no.JX187609), 15061 (NVDC-NM2, GenBank accession no.KC492504), 15058(NVDC-NM3, GenBank accession no.KC492505), and 15058(NVDC-FJ, GenBank accession no.KC492506) nucleotides(nt) in length excluding the 3′ poly(A) tails. The complete genome nucleotide homology of these four isolates was 85.6–95.5 %. They also shared 87.4–90.7 % nt homology with Lelystad PRRSV (the PRRSV-Type 1 reference strain), 86.9–88.8 % with the PRRSV-Type 1 live-attenuated vaccine, 72.3–78.5 % with Lena PRRSV (highly pathogenic PRRSV-Type 1 strain), and 53.2–54.5 % with VR-2332 PRRSV (the PRRSV-Type 2 prototype strain). In comparison to the genomic sequences of another two Chinese PRRSV-Type 1 isolates BJEU06-1 and NMEU09-1, these isolates shared 86.5–93.7 % homology with BJEU06-1 and 84.3–87.6 % homology with NMEU09-1. To further explore the genetic variety of these four isolates, the sequence of NVDC-NM1-2011 was compared to that of NVDC-NM2, NVDC-NM3, NVDC-FJ, LV, BJEU06-1, NMEU09-1, EuroPRRS, 01CB1, SD01-08, KNU-07, HKEU16, and Lena PRRSV (Table 1).
5′-UTR and 3′-UTR sequence variations of 4 PRRSV-Type 1 isolates
The 5′ terminus of the PRRSV genome contains a conserved start motif and the UUAACC transcription regulatory sequence (TRS) which are involved in transcription and replication [23–25]. In this study, the 5′-UTR region of all four isolates was 221 nucleotides in length. Moreover, the 12-nt 5′-leader start motif and the leader TRS were strictly conserved in the 5′-UTR of 4 PRRSV-Type 1 isolates (Fig. 1a). The 5′-UTR region of LV and VR-2332 strains were previously shown to contain three invariant nucleotide stretches just upstream of the leader TRS [24]. As expected, the nucleotide sequences of the three invariant motifs (Fig. 1a, solid boxes) were well maintained in NVDC-NM2, NVDC-NM3, and NVDC-FJ isolates. However, in the NVDC-NM1-2011 isolate C was substituted with U in the third stretch, a mutation similar to the 5′-UTR motifs changes observed in the BJEU06-1 isolate. The 3′-UTR of four isolates was 114 nucleotides in length and contained 5 or 9 nucleotide changes (Fig. 1b).

Alignment of 5′-UTR (A) and 3′-UTR (B) sequences of PRRSV-Type 1 viruses. a The12-nt start motif, leader TRS, and three invariant nucleotide stretches were indicated by a black box, a dotted line box, and solid boxes, respectively. b Alignment of 3′-UTR sequence of PRRSV-Type 1 viruses
Amino acid alteration in Nsp2 of 4 PRRSV-Type 1 isolates
The highly variable PRRSV protein, Nsp2, is the least conserved viral protein [2, 26]. In this study, several differences were found in the NSP2 aa sequence of all four isolates, when compared to the NSP2 sequence of the reference LV strain. Six aa deletions (deletion of 1 aa at residue 277, 4 aa at residue 357–360, and 1 aa at residue 411) were observed within the NSP2 of the NVDC-NM2, NVDC-NM3, and NVDC-FJ isolates, and five discontinuous deletions (4 aa deletions at residue 357–360 and 1 aa deletion at residue 411) were observed in the NVDC-NM1-2011 isolate (Fig. 2). Notably, in four isolates, 1 aa was deleted at residue 411, located in ES4 as not found in other viruses [27, 28]. In addition, a unique insertion of arginine was found at residue 757 of NVDC-NM2 that was not present in all the other viruses.

Alignment of NSP2 amino acid sequences of four isolates with other PRRSV-Type 1 strains. The amino acid discontinuous deletions detected in four isolates were indicated with solid boxes. One amino acid insertion at position 757 was indicated with a dotted line box. The previously identified B-cell epitope sites (ES) were indicated with underlining
Genetic characteristics of structural ORFs of 4 PRRSV-Type 1 isolates
The structural ORF nucleotide sequences (ORFs 2-7) of four isolates shared 91.4 % (NVDC-NM1-2011), 88.5 % (NVDC-NM2), 88.8 % (NVDC-NM3), and 88.7 % (NVDC-FJ) nucleotide homology with LV strain. The amino acid sequences of four isolates shared 83.7–96.1 % identity with the LV strain (Table 2).The structural proteins GP3 and GP5 were next analyzed with other representative PRRSV-Type 1 isolates from China and other countries.
GP3, a minor structural protein contains a hypervariable region (aa 237–252) in the overlap regions of ORF3 and ORF4 in PRRSV-Type 1 viruses [29]. Consistent with previous studies, the GP3 hypervariable region of four isolates contained multiple deletions and mutations. Continuous deletion of 8 aa (at positions 240–247 of GP3 or 60–67 of GP4) was observed in the hypervariable region of NVDC-NM2, NVDC-NM3, and NVDC-FJ isolates (Fig. 3a, b). In addition, 2 aa mutations (N27-T27 and S29-N29) were detected in one of seven potential N-glycosylation sites in GP3, indicated by the red boxes in Fig. 3a. In contrast, only 1 aa deletion (at position 245 of GP3 and position 65 of GP4) was detected in the GP3 hypervariable region of NVDC-NM1-2011 and no glycosylation site changes were found (Fig. 3a, b).

Amino acid variants in ORF3 and ORF4 of four isolates and other PRRSV-Type 1 viruses. a The 1-aa deletion at position 245 of ORF3 in NVDC-NM1-2011 and the 8-aa deletion at position 240–247 in ORF3 in the other three isolates were indicated by a solid box. Seven potential N-glycosylation sites in ORF3 were indicated by red boxes. b The 1-aa deletion at position 65 of ORF4 in NVDC-NM1-2011 and the 8-aa deletion at position 60–67 in ORF4 of the other 3 isolates were indicated by a solid box (Color figure online)
GP5, the most variable envelope glycoprotein, was encoded by ORF5 and comprised 201 amino acids in PRRSV-Type 1 viruses. This protein binds to M protein to form a disulfide-linked heterodimer that contains multiple primary neutralization sites [30, 31]. Previous studies indicated that the hypervariable region of GP5 includes the signal peptide sequence (at residues 1–33) and two ectodomains (at residues 33–67 and 89–109). Variation in these regions tends to affect the number of potential N-glycosylation sites and is expected to confer sensitivity to selective pressure from the porcine antibody response [32, 33]. In this study, we detected 11 amino acid substitutions within the ORF5 hypervariable region of NVDC-NM1-2011 (2 aa substitutions were found in the signal peptide sequence and 9 aa substitutions in two ectodomains) when compared to the LV PRRSV strain. 1 aa substitution (at residue 189) was found in ES13 (Fig. 4). In contrast, 17 amino acid substitutions were observed within the ORF5 hypervariable region of the 3 other isolates including substitution of 6 aa in the signal peptide sequence and 11 aa in two ectodomains. Notably, an additional N-glycosylation site at position 37 was detected in all four isolates (Fig. 4).

Amino acid variants in ORF5 of four isolates, compared with other PRRSV-Type 1 viruses. The functional domains were reported according to Stadejek et al. [33]. The putative signal sequences (dotted line boxes) and hypervariable domains (solid boxes) were indicated. Potential N-glycosylation sites were shown in the red boxes. The mutation at position 37 (D37 → N37) in all four isolates as shown by the red box. One mutation observed at ES13 of NVDC-NM1-2011 was indicated by the blue box (Color figure online)
Phylogenetic analysis of 4 PRRSV-Type 1 isolates and other PRRS viruses
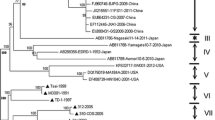
To establish genetic relationships between the isolated strains and previously characterized strains, phylogenetic analysis was performed on the full-length genomic sequences of the four isolates, 22 PRRSV strains including 5 PRRSV-Type 2 viral strains and 13 PRRSV-Type 1 viral strains in the GenBank database (Suppl. Table). The four isolates could be divided into different branches within the PRRSV-Type 1 Subtype 1 that is predominant in Europe [34]. NVDC-NM2, NVDC-NM3, and NVDC-FJ were closely related within the same branch, and NVDC-NM1-2011 fall into separate branch within Subtype 1 (Fig. 5). In addition, further phylogenetic analysis based on ORF5 and ORF7 nucleotide sequences also indicated that these isolates could be divided into two branches within the PRRSV-Type 1 Subtype 1 (data not shown).

Phylogenetic analysis of four isolates with other PRRSV-Type 1 strains based on the complete genomic sequence. Alignments were analyzed using the neighbor-joining method with the Kimura 2-parameter algorithm in MEGA4.1. Each isolate was indicated by virus name, GenBank accession no., and country of origin. The four isolates were indicated with dark triangle symbols. The reliability of the tree was assessed by bootstrap analysis with 1000 replications
Discussion
PRRS has now emerged as one of the most prevalent swine diseases in China, particularly after the unparalleled outbreaks of HP-PRRSV since 2006 [35, 36]. While PRRSV-Type 2 strains have previously been detected most often in China, the PRRSV-Type 1 has also been detected in China since 2011 [17]. In this study, nation-wide PRRSV surveillance of serum samples collected from 80 breeding swine farms provided the opportunity to comprehensively study the prevalence of PRRSV-Type 1 virus in China.
Four PRRSV-Type 1 viruses were successfully isolated and were subjected to genomic sequencing. Genomic sequence analysis confirmed the diversity of the PRRSV-Type 1 virus in China. In comparison to the LV strain, the complete genomic sequences of four isolates contained mutations within the nonstructural and structural proteins. Nsp1ß, Nsp2, ORF3, ORF4, and ORF5 exhibited the highest level of variation. PRRSV-Type 1 viruses were previously reported to contain different deletions within Nsp2 or ORF3 hypervariable regions than the LV strain [29–33]. In this study, all four isolates also contained deletions in these two hypervariable regions.
NSP2, the largest PRRSV protein, is crucial for the proteolytic processing of proteins during virus replication and modulation of immune responses [29, 30]. This protein possesses a putative conserved cysteine protease domain and contains six B-cell epitopes. ES2 is located in the putative cysteine protease domain, a domain important for viral survival and modulation of immune responses [28, 29]. In this study, ES2 was well conserved in the four isolates, which is consistent with earlier findings. The most variable region of NSP2 in these four isolates was located between the ES3 and ES4. Four aa deletions were detected in ES3 and 1 aa deletion was detected in ES4. Whether such deletions influence the virulence of PRRSV strains remains to be investigated.
The greatest amino acid variability in these isolates was detected in GP3 and GP5 in accordance with previous studies [31–33, 37, 38]. GP3 contains a hypervariable region located in the carboxyl-terminal end overlapping with ORF4. Previous studies indicated that this region may be subject to more rapid changes than other regions during immune selective pressure [29, 30, 38]. Moreover, the ES12 of GP4 is also located in this overlap region, and may act as a decoy epitope and play an important role in delaying the induction of PRRSV-neutralizing antibodies [17, 29, 38, 39]. In this study, the 8-aa deletion in NVDC-NM2, NVDC-NM3, and NVDC-FJ and 1 aa deletion of NVDC-NM1-2011 was located in ES12, which led to the disruption of the ES12 conformation.
GP5 is the most variable PRRSV structural protein. Amino acid variations in GP5 were mainly observed in the hypervariable region of the ectodomain, which possesses many potential N-glycosylation sites. These potential N-glycosylation sites are involved in viral immune evasion and minimize the virus-neutralizing antibody response [17, 29, 30, 32]. All four strains isolated in this study differed from LV at position 37 (D37-N37) and NVDC-NM1-2011 contained a 1 aa mutation in ES13. Whether the increase in glycosylation sites or amino acid mutation in GP3 and GP5 immune-dominant epitopes are related to the sensitivity of PRRSV to neutralization remains unknown.
To gain further insight into the genetic relationships between these isolates and other PRRSV-Type 1 isolates, phylogenetic analysis was performed on the whole genome of the four isolates, in addition to 22 PRRSV strains including 5 PRRSV-Type 2 viruses, and 13 PRRSV-Type 1 prototypic strains. We also performed phylogenetic analysis of the ORF5 of 100 strains and the ORF7 of 96 strains listed in the GenBank database. The phylogenetic trees revealed that all four isolates belonged to PRRSV-Type 1 Subtype 1, and could be further divided into two branches. NVDC-NM1-2011 belongs to one branch, while the other three strains were closely related within the other branch. Remarkably, NVDC-NM1-2011, NVDC-NM2, and NVDC-NM3 isolated from different breeding swine farms in the same province were divided into different branches. These results suggested that there was no clear geographical link between the four isolates, and suggested that independent EU PRRSV evolution is currently occurring in Chinese swine herds.
In summary, these results indicate that EU genotype PRRSV underwent gradual variation and has accumulated genomic changes since its introduction to Mainland China.
References
G. Wensvoort, C. Terpstra, J.M. Pol, E.A. terLaak, M. Bloemraad, E.P. deKluyver, C. Kragten, L. van Buiten, A. denBesten, F. Wagenaar, J.M. Broekhuijsen, P.L.J.M. Moonen, T. Zetstra, E.A. de Boer, H.J. Tibben, M.F. de Jong, P. van‘t Veld, G.J.R. Greenland, J.A. vanGennep, M.Th. Voets, J.H.M. Verheijden, J. Braamskamp, Vet. Q. 13(3), 121–130 (1991)
R. Allende, T.L. Lewis, Z. Lu, D.L. Rock, G.F. Kutish, A. Ali, A.R. Doster, F.A. Osorio, J. Gen. Virol. 80, 307–315 (1999)
J.J. Meulenberg, M.M. Hulst, E.J. de Meijer, P.L. Moonen, A. den Besten, E.P. de Kluyver, G. Wensvoort, R.J. Moormann, Virology 192, 62–72 (1993)
J.J. Meulenberg, A. Petersen-den Besten, E.P. De Kluyver, R.J. Moormann, W.M. Schaaper, G. Wensvoort, Virology 206, 155–163 (1995)
A.E. Firth, J.C. Zevenhoven-Dobbe, N.M. Wills, Y.Y. Go, U.B. Balasuriya, J.F. Atkins, E.J. Snijder, C.C. Posthuma, J. Gen. Virol. 92(Pt5), 1097–1106 (2011)
C.R. Johnson, T.F. Griggs, J. Gnanandarajah, M.P. Murtaugh, J. Gen. Virol. 92, 1107–1116 (2011)
E.J. Snijder, J.J. Meulenberg, J. Gen. Virol. 79, 961–979 (1998)
M.B. Oleksiewicz, A. Bøtner, K.G. Madsen, T. Storgaard, Vet. Microbiol. 64(1), 7–22 (1998)
L. Zhou, S. Chen, J. Zhang, J. Zeng, X. Guo, X. Ge, D. Zhang, H. Yang, Virus Res. 145(1), 97–105 (2009)
J.E. Collins, D.A. Benfield, W.T. Christianson, L. Harris, J.C. Hennings, D.P. Shaw, S.M. Goyal, S. McCullough, R.B. Morrison, H.S. Joo, D. Gorcyca, D. Chladek, J. Vet. Diagn. Invest. 4, 117–126 (1992)
K.G. Madsen, C.M. Hansen, E.S. Madsen, B. Strandbygaard, A. Bøtner, K.J. Sørensen, Arch. Virol. 143(9), 1683–1700 (1998)
C. Egli, B.L. Liu, M.A. Hofmann, J. Virol. Methods 98(1), 63–75 (2001)
B. Guo, Z. Chen, W. Liu, Y. Cui, Chin. J. Anim. Poult. Infect. Dis. 17, 1–4 (1996)
A. Amonsin, R. Kedkovid, S. Puranaveja, P. Wongyanin, S. Suradhat, R. Thanawongnuwech, Virol. J. 6, 143 (2009)
G. Balka, A. Hornyak, A. Balint, I. Kiss, S. Kecskemeti, T. Bakonyi, M. Rusvai, Vet. Microbiol. 127, 128–135 (2008)
A. Bøtner, B. Strandbygaard, K.J. Sørensen, P. Have, K.G. Madsen, E.S. Madsen, S. Alexandersen, Vet. Rec. 141, 497–499 (1997)
N.H. Chen, Z. Cao, X.L. Yu, X.Y. Deng, T. Zhao, L. Wang, Q. Liu, X. Li, K. Tian, J. Gen. Virol. 92, 880–892 (2011)
S.B. Kleiboeker, S.K. Schommer, S.M. Lee, S. Watkins, W. Chittick, D.J. Polson, Vet. Diagn. Invest. 17, 165–170 (2005)
H.S. Kim, J. Kwang, I.J. Yoon, H.S. Joo, M.L. Frey, Arch. Virol. 133, 477–483 (1993)
D. Zeman, R. Neiger, M. Yaeger, E. Nelson, D. Benfield, P. Leslie-Steen, J. Thomson, D. Miskimins, R. Daly, M. Minehart, J. Vet. Diagn. Invest. 5(4), 522–528 (1993)
G. Lu, E.N. Moriyama, Brief Bioinform. 5(4), 378–388 (2004)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24, 1596–1599 (2007)
V. Kapur, M.R. Elam, T.M. Pawlovich, M. Murtaugh, J. Gen. Virol. 77, 1271–1276 (1996)
M.B. Oleksiewicz, A. Bøtner, J. Nielsen, T. Storgaard, Arch. Virol. 144, 981–987 (1999)
G. van Marle, J.C. Dobbe, A.P. Gultyaev, W. Luytjes, W.J. Spaan, E.J. Snijder, Proc. Natl. Acad. Sci. USA 96(21), 12056–12061 (1999)
Y. Fang, D.Y. Kim, S. Ropp, P. Steen, J. Christopher-Hennings, E.A. Nelson, R.R. Rowland, Virus Res. 100, 229–235 (2004)
M. de Lima, A.K. Pattnaik, E.F. Flores, F.A. Osorio, Virology 353, 410–421 (2006)
M.B. Oleksiewicz, A. Bøtner, P. Toft, P. Normann, T. Storgaard, J. Virol. 75, 3277–3290 (2001)
S.L. Ropp, C.E. Wees, Y. Fang, E.A. Nelson, K.D. Rossow, M. Bien, B. Arndt, S. Preszler, P. Steen, J. Christopher-Hennings, J.E. Collins, D.A. Benfield, K.S. Faaberg, J. Virol. 78(7), 3684–3703 (2004)
S. Dea, C.A. Gagnon, H. Mardassi, B. Pirzadeh, D. Rogan, Arch. Virol. 145, 659–688 (2000)
M.H. Verheije, T.J. Welting, H.T. Jansen, P.J. Rottier, J.J. Meulenberg, Virology 30, 364–373 (2002)
B. Pirzadeh, C.A. Gagnon, S. Dea, Can. J. Vet. Res. 62, 170–177 (1998)
T. Stadejek, A. Stankevicius, T. Storgaard, M.B. Oleksiewicz, S. Belak, T.W. Drew, Z. Pejsak, J. Gen. Virol. 83, 1861–1873 (2002)
T. Stadejek, M.B. Oleksiewicz, A.V. Scherbakov, A.M. Timina, J.S. Krabbe, K. Chabros, D. Potapchuk, Arch. Virol. 153, 1479–1488 (2008)
K.G. Tian, X.L. Yu, T.Z. Zhao, Y.J. Feng, Z. Cao, C. Wang, Y. Hu, X. Chen, D. Hu, X. Tian, D. Liu, S. Zhang, X. Deng, Y. Ding, L. Yang, Y. Zhang, H. Xiao, M. Qiao, B. Wang, L. Hou, X. Wang, X. Yang, L. Kang, M. Sun, P. Jin, S. Wang, Y. Kitamura, J. Yan, G.F. Gao, PLoS One 2(6), e526 (2007)
T.Q. An, Z.J. Tian, C.L. Leng, J.M. Peng, G.Z. Tong, Emerg. Infect. Dis. 17(9), 1782–1784 (2011)
R. Forsberg, M.B. Oleksiewicz, A.M.K. Petersen, J. Hein, A. Bøtner, T. Storgaard, Virology 289, 174–179 (2001)
M.B. Oleksiewicz, A. Bøtner, P. Toft, T. Grubbe, J. Nielsen, S. Kamstrup, T. Storgaard, Virology 267, 135–140 (2000)
T.W. Drew, J.P. Lowings, F. Yapp, Vet. Microbiol. 55, 209–221 (1997)
Acknowledgments
This work was supported by Grants from the Swine Industry Technology System Building of Beijing (GWZJ-2009-05), the National Basic Research Program of China (2008FY130100-2), and the Scientific Achievement Transformation Program (2009GB23260435). We thank Dr. Xiangdong Li at National Research Center for Veterinary Medicine, Cuiwei Road, High-Tech. District, Luoyang, Henan Province, China for providing revision of the grammar, and readability of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by William Dundon.
Zhi Zhou and Qi Liu are contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhou, Z., Liu, Q., Hu, D. et al. Complete genomic characterization and genetic diversity of four European genotype porcine reproductive and respiratory syndrome virus isolates from China in 2011. Virus Genes 51, 375–384 (2015). https://doi.org/10.1007/s11262-015-1256-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1256-z