
Abstract
The H4 subtype of the influenza virus was first isolated in 1999 from pigs with pneumonia in Canada. H4 avian influenza viruses (AIVs) are able to cross the species barrier to infect humans. In order to better understand the genetic relationships between H4 AIV strains circulating in Eastern China and other AIV strains from Asia, a survey of domestic ducks in live poultry markets was undertaken in Zhejiang province from 2013 to 2014. In this study, 23 H4N2 (n = 14) and H4N6 (n = 9) strains were isolated from domestic ducks, and all eight gene segments of these strains were sequenced and compared to reference AIV strains available in GenBank. The isolated strains clustered primarily within the Eurasian lineage. No mutations associated with adaption to mammalian hosts or drug resistance was observed. The H4 reassortant strains were found to be of low pathogenicity in mice and able to replicate in the lung of the mice without prior adaptation. Continued surveillance is required, given the important role of domestic ducks in reassortment events leading to new AIVs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Based on the antigenic properties of the hemagglutinin (HA) and neuraminidase (NA) glycoproteins, influenza A viruses are classified into 18 HA and 11 NA subtypes [1, 2]. Domestic ducks are known to harbor most of the avian influenza viruses (AIVs) subtypes, and are considered a natural reservoir for AIVs [3]. While AIV infection is usually asymptomatic in domestic ducks, they provide an environment for the reassortment of low pathogenic avian influenza (LPAI) viruses, which can serve as progenitors of highly pathogenic avian influenza (HPAI) viruses [4, 5]. Since 1997, the H5, H7, and H9 AIV subtypes have caused disease outbreaks in poultry worldwide and have infected humans in many countries [6–11]. However, there is relatively little information regarding the molecular characteristics of other AIV subtypes in Eastern China, including the H4 subtype.
The AIV H4 subtype infects domestic ducks and is known to circulate in live poultry markets (LPMs) in China [4, 12]. H4 viruses were also isolated in 1999 from pigs with pneumonia in Canada [13], and have been transmitted to pig populations in southeastern China [14]. Furthermore, H4 AIVs are able to cross the species barrier to infect humans [15, 16]. LPMs are considered a major source of AIV dissemination, and sites for potential AIV reassortment and interspecies transfers [4, 5]. Given these factors, active surveillance of the AIVs circulating in LPMs should be used in an early warning system for AIV outbreaks.
To better understand the genetic relationships between the H4 strains from Eastern China and other H4 strains from throughout Asia, a survey of the H4 AIV subtypes circulating in Zhejiang province, Eastern China, from 2013 to 2014 was performed. The gene segments of the isolated field strains were sequenced and compared with reference sequences available in GenBank.
Materials and methods
Cloacal swabs were collected from apparently healthy ducks in LPMs in Zhejiang province, Eastern China, from March 2013 to December 2014. Viruses were isolated from the duck cloacal swab material by inoculation into embryonated chicken eggs as previously described [17]. After incubation at 37 °C for 72 h, the allantoic fluid was harvested and tested by HA assay. Specific pathogen-free 1 % chicken red blood cells were prepared. HA titers were then determined by adding 50 µL of the 1 % chicken red blood cells in phosphate-buffered saline (PBS) to 50 µL of a two-fold serial dilution of virus in 96 ‘V’-well microtiter plates. The microtiter plates were incubated for 30 min at 25 °C. HA titers were defined as the reciprocal values of the highest dilutions that caused complete hemagglutination. Viruses in the HA-positive samples were subtyped by reverse transcription-PCR (RT-PCR) using specific primers [17]. Aliquots of the allantoic fluid containing viruses were stored as stocks at −80 °C for further analysis.
RNA was extracted from the HA-positive allantoic fluid using a viral RNA mini kit (Qiagen, Germany) according to the manufacturer’s instructions. Reverse transcription was performed using the Uni12 primer: 5′-AGCAAAAGCAGG-3′. RT-PCR was performed using a One-Step RNA PCR Kit (TaKaRa, China). All of the viral gene segments were amplified with segment-specific primers as described previously [18]. The fragments were sequenced using a BigDye Terminator V.3.0 Cycle Sequencing Ready Reaction kit (Life Technologies, USA), according to the manufacturer’s instructions. The nucleotide sequences were deposited into GenBank under the accession numbers KT589138-321. The sequences were analyzed using the BioEdit version 7.0.9.0 DNA analysis software. Phylogenetic trees were constructed using the molecular evolutionary genetics analysis (MEGA) software version 5.05, applying the maximum likelihood method and Tamura-Nei model with bootstrap analysis (1000 replicates) [19]. The sequences of the reference strains used in this study were obtained from the Influenza Sequences Database (http://www.ncbi.nlm.nih.gov).
To evaluate the ability of each virus to replicate in vivo and assess its pathogenicity in mice, 15 6-week-old female BALB/c mice were intranasaly inoculated with 106.0 EID50 of virus in a 0.05-mL volume of phosphate-buffered saline. Three (3) mice were sacrificed at 3, 6, and 9 days post-inoculation (dpi). The lung, brain, heart, kidney, spleen, and liver were harvested from each mouse, and homogenized in 1 mL of cold PBS. The samples were centrifuged at 2500g for 5 min. Embryonated chicken eggs were used to determine the EID50s of the supernatants using the method of Reed and Muench [20]. The remaining six mice were observed to determine survival for 14 dpi as described previously [21]. The animal studies were carried out in accordance with the recommendations of the Office International des Epizooties (OIE) [22] and approved by the First Affiliated Hospital, School of Medicine, Zhejiang University (2015-015).
Results
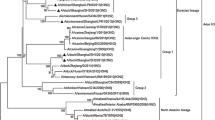
A total of 3210 cloacal swabs were collected from apparently healthy ducks in LPMs during the course of the survey. One hundred and nine strains were isolated, and 10 HA subtypes (H1, H2, H3, H4, H5, H6, H7, H9, H10, and H11), eight NA subtypes (N1, N2, N3, N4, N6, N7, N8, and N9), and 21 AIV subtypes were identified (Table S1). Of these, 23 (0.72 %) were H4 AIV isolates. The H4 isolates were either H4N2 (n = 14) or H4N6 (n = 9). The phylogenetic analysis of the gene sequences for HA, NA, PB2, PB1, PA, NP, M, and NS, indicated that the H4 AIVs were clustered in the Eurasian lineage. The phylogenetic trees for HA (Fig. 1), N6 (Fig. 2), and N2 (Fig. 3) are shown, while the phylogenetic trees for the remaining genes are presented in supplementary data (Fig. S1). The HA phylogenetic tree suggested that three different H4 genetic groups were co-circulating in Zhejiang province. The N6 phylogenetic tree suggested that the H4N6 AIVs had a high degree of nucleotide similarity to H4N6 viruses isolated in Eastern Asia from 2008 to 2012. The tree also indicated that the H4N6 viruses in this study and the novel 2014 H5N6 influenza virus, which infected humans, had different ancestral N6 genes. The N2 phylogenetic tree showed that the NA in the H4N2 viruses was derived from the H3N2 viruses’ epidemic in Asian birds since 2009. The remaining phylogenetic trees suggest that the internal genes (PB2, PB1, PA, NP, M, and NS) also belong to the Eurasian lineage (Fig. S1). Furthermore, they had a high degree of nucleotide similarity to different H4 AIV subtypes from aquatic birds and poultry throughout Eastern Asia, in particular domestic ducks from Eastern China.

Phylogenetic tree based on the nucleotide sequence of hemagglutinin (HA) (positions 1-1695) comparing the isolated H4 avian influenza viruses (AIVs) isolated in this study to reference AIV sequences published in GenBank. The tree was created by the maximum likelihood method and bootstrapped with 1000 replicates using the MEGA5 software version 5.05. The H4N2 and H4N6 viruses characterized are highlighted by a triangle. The scale bar represents the distance unit between sequence pairs

Phylogenetic tree based on the nucleotide sequence of neuraminidase (NA) subtype 6 (N6; positions 1-1413) gene sequence comparing the isolated H4N6 avian influenza viruses (AIVs) isolated in this study to reference AIV sequences published in GenBank. The tree was created by the maximum likelihood method and bootstrapped with 1000 replicates using the MEGA5 software version 5.05. The H4N6 viruses characterized are highlighted by a triangle, and the novel 2014 H5N6 influenza virus that caused human infection is highlighted by a dot. The scale bar represents the distance unit between sequence pairs

Phylogenetic tree based on the nucleotide sequence of neuraminidase (NA) subtype 2 (N2; positions 1-1410) gene sequence comparing the isolated H4N2 avian influenza viruses (AIVs) isolated in this study to reference AIV sequences published in GenBank. The tree was created by the maximum likelihood method and bootstrapped with 1000 replicates using the MEGA5 software version 5.05. The H4N2 viruses characterized are highlighted by a triangle. The scale bar represents the distance unit between sequence pairs
Two representative isolates A/duck/Zhejiang/727145/2014 (H4N2; ZJ-727145) and A/duck/Zhejiang/D15/2013 (H4N6; ZJ-D15) were chosen for more in-depth analysis. The percent sequence similarity for each gene segment in ZJ-727145 (H4N2) and ZJ-D15 (H4N6) compared with their closest genetic relative is shown in Table 1. The HA genes of ZJ-727145 (H4N2) and ZJ-D15 (H4N6) had the highest nucleotide similarity to A/duck/Mongolia/OIE-7438/2011 (H4N6). The NA genes of ZJ-727145 (H4N2) and ZJ-D15 (H4N6) showed the highest nucleotide similarity to A/environment/Hunan/S4304/2011 (H3N2) and A/duck/Zhejiang/D1-3/2013 (H3N6), respectively. The internal genes (PB2, PB1, PA, NP, M, and NS) of ZJ-727145 and ZJ-D15 shared the highest similarity with H1, H3, and H7 isolates, such as A/duck/Zhejiang/473/2013 (H1N4), A/duck/Huzhou/4227/2013 (H7N7), and A/duck/Shanghai/SH3/2013 (H3N2) (Table 1; Fig. 4). ZJ-727145 and ZJ-D15 are both reassortment viruses derived from multiple AIV subtypes from aquatic birds and poultry in Eastern Asia. This finding indicated that a reassortment event between H4 and H3 viruses occurred in these ducks, and provided additional evidence for the active evolution and segment reassortment H4 AIV subtypes in China.

A schematic representation of the putative genomic compositions of two representative H4 avian influenza viruses isolated from domestic ducks in Eastern China and their possible parent viruses. The eight gene segments (from top to bottom) in each virus are PB2, PB1, PA, HA, NP, NA, M, and NS. Each color represents a separate virus background. 775(H7N2), A/duck/Wenzhou/775/2013 (H7N2); 559(H7N7), A/pigeon/Wenzhou/559/2013 (H7N7); 4227(H7N7), A/duck/Huzhou/4227/2013 (H7N7); 7438(H4N6), A/duck/Mongolia/OIE-7438/2011 (H4N6); 473(H1N4), A/duck/Zhejiang/473/2013 (H1N4); S4304(H3N2), A/environment/Hunan/S4304/2011 (H3N2); RG3(H7N3), A/mallard/Republic of Georgia/3/2010 (H7N3); LBM48(H3N2), A/duck/Vietnam/LBM48/2011 (H3N2); ZJ2(H7N3), A/duck/Zhejiang/2/2011 (H7N3); D1-3(H3N6), A/duck/Zhejiang/D1-3/2013 (H3N6); 0607-13(H1N2), A/duck/Zhejiang/0607-13/2011 (H1N2); SH3(H3N2), A/duck/Shanghai/SH3/2013 (H3N2). The simplified schematic illustration is based on nucleotide-distance comparison and phylogenetic analysis (Color figure online)
Based on the deduced amino acid sequences of the HA gene, the HA cleavage site pattern PEKASR/GL occurs in all 23 H4 AIVs. The cleavage site pattern was consistent with a monobasic cleavage site and indicated that these strains were LPAI viruses. Amino acids at receptor binding sites were highly conserved (Fig. S2). The receptor binding sites in the isolated H4 AIVs were Q226 and G228, which were similar to the H4 AIVs in both the Eurasian and North American lineages. Thus, suggesting that this strain would preferentially bind to alpha 2–3-linked sialic acid receptors that are predominant in avian species [15, 23]. The L226 mutation that can increase affinity toward alpha 2–6-linked sialic acid receptors [15] was not observed in the isolated H4 AIVs.
HA glycosylation is associated with virulence and virus affinity for the influenza virus receptor [24, 25]. The specific polypeptide for N-linked glycosylation is Asn-X-Ser/Thr, where X can be any amino acid, except proline or aspartic acid [26]. Six (6) potential HA glycosylation sites at positions 14, 18, 34, 178, 310, and 497 were detected in the strains identified in this study. Interestingly, some of the H4 strains characterized here lacked the potential glycosylation sites at positions 14 and 18 (Fig. S2). This was also the case with other H4 subtype AIVs. It is unclear whether this alters viral affinity for the influenza receptor in these strains.
No mutations associated with resistance to NA inhibitors (oseltamivir and zanamivir, His274Tyr) or amantadines (Val27Ala and Ser31Asn) were observed in the NA and M2 proteins, respectively. Previous reports have shown that the mutation Glu627Lys in PB2 is associated with pathogenicity of H5N1 AIVs in mice [27]. Additional mutations such as Thr271Ala, Asp701Asn, and Ser714Arg contribute to enhanced influenza polymerase activity in mammalian host cells [28, 29]. However, neither of these types of mutations were observed in PB2 from the isolated H4 AIVs.
To test the in vivo pathogenicity of ZJ-727145 and ZJ-D15 in a mammalian host, mice were infected intranasally with 106.0 EID50 of each virus (Table 2). ZJ-727145 and ZJ-D15 were able to replicate without prior adaptation in mice. At 6 dpi, higher titers of ZJ-727145 and ZJ-D15 were detected in the lungs than at 3 dpi, but the viruses could not replicate in the brain, heart, liver, kidney, or spleen of the mice. The mice had survival rates of 100 % (6/6) up to 14 dpi. Taken together, these results indicate that these viruses are of low pathogenicity in mice.
Discussion
Several recent epidemiological studies of AIVs in China have found that domestic ducks in Central and Southern China are important to the genesis and evolution of AIV [12, 30, 31]. However, there is limited data about the molecular characteristics of H4 AIV in Eastern China. Zhejiang province is located in Eastern China and is an important location for migrating wild birds in East Asia. The H4 AIV strains isolated from domestic ducks in this study clustered with other Eurasian influenza viruses based on all eight gene segments. Our results also suggest that a reassortment event between H4 and H3 AIVs occurred in domestic ducks. The deduced amino acid sequences of the strains isolated here did not contain molecular markers linked to increased virulence in chickens and mammals, and no adaptations for drug resistance were identified in the HA, NA, M, or PB2 proteins. While the addition of multiple amino acids to the HA cleavage site, such as arginine (R) or lysine (K), can convert an LPAI into a HPAI subtype (H5 or H7) [32], these mutations were not observed. Although these strains were found to be of low pathogenicity in mice, they were able to replicate in the lung of the mice without prior adaptation. Our results were consistent with Zhang et al., [33] who showed that a duck H4N2 virus did not cause death in chickens or mice.
Since March 2013 a novel H7N9 virus associated with human deaths has emerged in Eastern China. Some investigators have suggested that the virus is a reassortant of H7N9 and H9N2 AIVs, and that the H7 and H9 viruses have been co-circulating widely in birds (especially domestic ducks) in Eastern China for several years [10, 34]. Our previous studies showed that reassortment events between multiple AIV subtypes (H1, H2, H3, H5, H7, and H10) have occurred in domestic ducks in LPMs in Zhejiang province since 2011 [17, 35–38]. Given that reassorted H4N2 and H4N6 viruses were identified in domestic ducks in this study, it is likely that domestic ducks play an important role in reassortment events leading to new AIVs. LPMs are considered a major source of influenza virus dissemination and potentially for influenza virus reassortment. Continued surveillance of domestic ducks in LPMs should be used as an early warning system for AIV outbreaks in poultry and humans.
References
X. Zhu, W. Yu, R. McBride, Y. Li, L.M. Chen, R.O. Donis, S. Tong, J.C. Paulson, I.A. Wilson, Proc. Natl. Acad. Sci. USA 110, 1458–1463 (2013)
S. Tong, X. Zhu, Y. Li, M. Shi, J. Zhang, M. Bourgeois, H. Yang, X. Chen, S. Recuenco, J. Gomez, L.M. Chen, A. Johnson, Y. Tao, C. Dreyfus, W. Yu, R. McBride, P.J. Carney, A.T. Gilbert, J. Chang, Z. Guo, C.T. Davis, J.C. Paulson, J. Stevens, C.E. Rupprecht, E.C. Holmes, I.A. Wilson, R.O. Donis, PLoS Pathog. 9, e1003657 (2013)
Y. Kawaoka, T.M. Chambers, W.L. Sladen, R.G. Webster, Virology 163, 247–250 (1988)
M. Liu, S. He, D. Walker, N. Zhou, D.R. Perez, B. Mo, F. Li, X. Huang, R.G. Webster, R.J. Webby, Virology 305, 267–275 (2003)
C. Cardona, K. Yee, T. Carpenter, Poult. Sci. 88, 856–859 (2009)
K.S. Li, Y. Guan, J. Wang, G.J. Smith, K.M. Xu, L. Duan, A.P. Rahardjo, P. Puthavathana, C. Buranathai, T.D. Nguyen, A.T. Estoepangestie, A. Chaisingh, P. Auewarakul, H.T. Long, N.T. Hanh, R.J. Webby, L.L. Poon, H. Chen, K.F. Shortridge, K.Y. Yuen, R.G. Webster, J.S. Peiris, Nature 430, 209–213 (2004)
M. Koopmans, B. Wilbrink, M. Conyn, G. Natrop, H. van der Nat, H. Vennema, A. Meijer, J. van Steenbergen, R. Fouchier, A. Osterhaus, A. Bosman, Lancet 363, 587–593 (2004)
D.M. Skowronski, S.A. Tweed, M. Petric, T. Booth, Y. Li, T. Tam, J Infect Dis 193, 899–900 (2006). author reply 900–891
K.M. Butt, G.J. Smith, H. Chen, L.J. Zhang, Y.H. Leung, K.M. Xu, W. Lim, R.G. Webster, K.Y. Yuen, J.S. Peiris, Y. Guan, J. Clin. Microbiol. 43, 5760–5767 (2005)
Y. Chen, W. Liang, S. Yang, N. Wu, H. Gao, J. Sheng, H. Yao, J. Wo, Q. Fang, D. Cui, Y. Li, X. Yao, Y. Zhang, H. Wu, S. Zheng, H. Diao, S. Xia, K.H. Chan, H.W. Tsoi, J.L. Teng, W. Song, P. Wang, S.Y. Lau, M. Zheng, J.F. Chan, K.K. To, H. Chen, L. Li, K.Y. Yuen, Lancet 381, 1916–1925 (2013)
K.T. Eames, C. Webb, K. Thomas, J. Smith, R. Salmon, J.M. Temple, BMC Infect. Dis. 10, 141 (2010)
A. Wu, Z. Xie, L. Xie, S. Luo, X. Deng, L. Huang, J. Huang, T. Zeng, Genome Announc. (2014). doi:10.1128/genomeA.00894-14
A.I. Karasin, I.H. Brown, S. Carman, C.W. Olsen, J. Virol. 74, 9322–9327 (2000)
A. Ninomiya, A. Takada, K. Okazaki, K.F. Shortridge, H. Kida, Vet. Microbiol. 88, 107–114 (2002)
A.C. Bateman, M.G. Busch, A.I. Karasin, N. Bovin, C.W. Olsen, J. Virol. 82, 8204–8209 (2008)
G. Kayali, E. Barbour, G. Dbaibo, C. Tabet, M. Saade, H.A. Shaib, J. Debeauchamp, R.J. Webby, PLoS ONE 6, e26818 (2011)
H.B. Wu, C.T. Guo, R.F. Lu, L.H. Xu, E.K. Wo, J.B. You, Y.T. Wang, Q.G. Wang, N.P. Wu, Virus Genes. 44, 441–449 (2012)
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Perez, Arch. Virol. 146, 2275–2289 (2001)
K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei, S. Kumar, Mol. Biol. Evol. 28, 2731–2739 (2011)
L. Reed, H. Muench, Am. J. Hyg. 27, 493–497 (1938)
W. Hai-bo, G. Chao-tan, L. Ru-feng, X. Li-hua, W. En-kang, Y. Jin-biao, W. Yi-ting, W. Qiao-gang, W. Nan-ping, Arch. Virol. 157, 1131–1136 (2012)
B. Olsen, V.J. Munster, A. Wallensten, J. Waldenstrom, A.D. Osterhaus, R.A. Fouchier, Science 312, 384–388 (2006)
T. Wisedchanwet, M. Wongphatcharachai, S. Boonyapisitsopa, N. Bunpapong, P. Kitikoon, A. Amonsin, Virol. J. 8, 131 (2011)
Y. Zhang, J. Zhu, Y. Li, K.C. Bradley, J. Cao, H. Chen, M. Jin, H. Zhou, PLoS ONE 8, e61397 (2013)
M. Iqbal, S.C. Essen, H. Xiao, S.M. Brookes, I.H. Brown, J.W. McCauley, Virology 433, 282–295 (2012)
A. Helenius, M. Aebi, Annu. Rev. Biochem. 73, 1019–1049 (2004)
K.A. Schat, J. Bingham, J.M. Butler, L.M. Chen, S. Lowther, T.M. Crowley, R.J. Moore, R.O. Donis, J.W. Lowenthal, PLoS ONE 7, e30960 (2012)
K.A. Bussey, T.L. Bousse, E.A. Desmet, B. Kim, T. Takimoto, J. Virol. 84, 4395–4406 (2010)
V. Czudai-Matwich, A. Otte, M. Matrosovich, G. Gabriel, H.D. Klenk, J. Virol. 88, 8735–8742 (2014)
G. Deng, D. Tan, J. Shi, P. Cui, Y. Jiang, L. Liu, G. Tian, Y. Kawaoka, C. Li, H. Chen, J. Virol. 87, 9452–9462 (2013)
K. Huang, H. Zhu, X. Fan, J. Wang, C.L. Cheung, L. Duan, W. Hong, Y. Liu, L. Li, D.K. Smith, H. Chen, R.G. Webster, R.J. Webby, M. Peiris, Y. Guan, J. Virol. 86, 6075–6083 (2012)
T. Horimoto, T. Ito, D.J. Alexander, Y. Kawaoka, J. Vet. Med. Sci. 57, 927–930 (1995)
H. Zhang, Q. Chen, Z. Chen, Virus Genes. 44, 24–31 (2012)
T.T. Lam, J. Wang, Y. Shen, B. Zhou, L. Duan, C.L. Cheung, C. Ma, S.J. Lycett, C.Y. Leung, X. Chen, L. Li, W. Hong, Y. Chai, L. Zhou, H. Liang, Z. Ou, Y. Liu, A. Farooqui, D.J. Kelvin, L.L. Poon, D.K. Smith, O.G. Pybus, G.M. Leung, Y. Shu, R.G. Webster, R.J. Webby, J.S. Peiris, A. Rambaut, H. Zhu, Y. Guan, Nature 502, 241–244 (2013)
W. Hai-bo, L. Ru-feng, W. En-kang, Y. Jin-biao, W. Yi-ting, W. Qiao-gang, X. Li-hua, W. Nan-ping, G. Chao-tan, Arch. Virol. 157, 2017–2021 (2012)
H. Wu, R. Lu, X. Wu, X. Peng, L. Xu, L. Cheng, X. Lu, C. Jin, T. Xie, H. Yao, N. Wu, Infect. Genet. Evol. 29, 1–5 (2015)
H. Wu, X. Peng, L. Xu, C. Jin, L. Cheng, X. Lu, T. Xie, H. Yao, N. Wu, Arch. Virol. 159, 3377–3383 (2014)
X. Peng, H. Wu, C. Jin, H. Yao, X. Lu, L. Cheng, N. Wu, Virus Genes. 48, 391–396 (2014)
Acknowledgments
This study was supported by Grants from the National Key Technologies R&D Programme for the 12th Five-Year Plan of China (2012ZX1000-004-005), National Science Foundation of the People’s Republic of China (81502852), Zhejiang Provincial Natural Science Foundation of China (Y15H190006), and the Independent Task of State Key Laboratory for Diagnosis and Treatment of Infectious Diseases (Nos. 2014ZZ12 and 2015ZZ05).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by William Dundon.
Haibo Wu and Xiuming Peng contributed equally to the work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11262_2015_1245_MOESM1_ESM.doc
Figure S1. Phylogenetic trees based on the gene sequences of PB2 (positions 1-2280), PB1 (positions 1-2274), PA (positions 1-2151), NP (positions 1-1490), M (positions 1-924), and NS (positions 1-838) for the natural reassortant H4 avian influenza virus (AIV) and reference AIV strains. The tree was created by the maximum likelihood method and bootstrapped with 1000 replicates using the MEGA5 software version 5.05. The H4N2 and H4N6 viruses isolated in this study are highlighted by a triangle. The scale bar represents the distance unit between sequence pairs. Supplementary material 1 (DOC 970 kb)
11262_2015_1245_MOESM2_ESM.doc
Figure S2. Comparison of deduced HA amino acid sequences of the H4 avian influenza viruses (AIVs) isolated in this study and reference AIV strains using the MegAlign program. Identical amino acids are shown as dots. The blue box represents the HA sequence at the cleavage site, black boxes represent potential N-linked glycosylation sites, and red/yellow boxes indicate residues involved in the receptor-binding site. Supplementary material 2 (DOC 2091 kb)
Rights and permissions
About this article

Cite this article
Wu, H., Peng, X., Peng, X. et al. Genetic characterization of natural reassortant H4 subtype avian influenza viruses isolated from domestic ducks in Zhejiang province in China from 2013 to 2014. Virus Genes 51, 347–355 (2015). https://doi.org/10.1007/s11262-015-1245-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-015-1245-2