
Abstract
Nine avian influenza A viruses (AIVs), H1N2 (n = 2) and H1N3 (n = 7), were isolated from domestic ducks in live poultry markets in Zhejiang Province, Eastern China, in 2011. All viruses were characterized by whole genome sequencing with subsequent phylogenetic analysis and genetic comparison. Phylogenetic analysis of all eight viral genes showed that the viruses clustered in the Eurasian lineage of influenza A viruses. The hemagglutinin cleavage site of all viruses displayed features of a monobasic cleavage site. Although there was no evidence of re-assortment in subtype H1 AIVs among the avian species and mammalian hosts in this study, continued surveillance is needed considering the important role of the domestic duck in the dissemination and re-assortment of AIVs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Avian influenza A viruses (AIVs) are members of the Orthomyxoviridae family and contain eight segments of single-stranded RNA with negative polarity [1]. AIVs are classified into subtypes based on their envelope proteins, hemagglutinin (HA) and neuraminidase (NA). Currently, all 16 the HA and the 9 NA subtypes were isolated from aquatic birds, such as ducks and geese, which were considered the natural reservoir of AIVs [2]. Though aquatic birds did not display the symptoms when they were infected with AIVs, they provide an environment for the re-assortment of low pathogenic avian influenza (LPAI) viruses which can serve as progenitors of highly pathogenic avian influenza (HPAI) viruses [3, 4].
Live poultry markets (LPMs) are where domestic poultry are slaughtered and sold to households in China and other countries [3, 5, 6]. LPMs are considered a major source of AIV dissemination and sites for potential influenza virus re-assortment as well as interspecies transfers [4, 7, 8]. Previous studies have shown that LPMs in Hong Kong were closely linked to the infection of humans with H5N1 AIVs in 1997 and H9N2 AIVs in 1999 [9–11]. Since LPMs play an important role in the dissemination of AIVs, active surveillance of HPAI and LPAI viruses in LPMs should be used in an early warning system for avian influenza outbreaks [12–14].
During the surveillance of poultry for AIVs in LPMs in Zhejiang Province, Eastern China, in 2011, H1N2 (n = 2) and H1N3 (n = 7) AIVs were isolated from apparently healthy domestic ducks. Whole genome sequencing and phylogenetic analyses of the viruses were performed. The results suggest that these viruses have re-assortment between HPAI and LPAI viruses in avian species, and continued circulation of these viruses may bear threats for both avian species and humans.
Materials and methods
Virus isolation
One hundred cloacal swabs were collected from apparently healthy Shaoxing pockmark duck (Anas platyrhyncha var. domestica, from commercial farms), in LPMs (n = 2) in Hangzhou, Zhejiang Province, January to June, in 2011. Each swab was eluted with 2.0 ml phosphate-buffered saline containing 0.2% bovine serum albumin (BSA), 4 × 106 U/l penicillin G, and 400 mg/l streptomycin sulfate. The samples were sterilized using a 0.22-μm filter and inoculated into the allantoic cavities of 9-day-old specific-pathogen-free embryonated eggs. After incubation at 37°C for 72 h, the allantoic fluid was harvested and tested by hemagglutination (HA) assay [15]. The subtypes of the virus isolates were determined by conventional HA inhibition (HI) and NA inhibition (NI) assays, according to the Office International des Epizootics (OIE) manual [16]. Aliquots of virus allantoic fluid stock were stored at −80°C before used. All the experiments with the AIVs were performed in a Biosafety Level 3 laboratory.
RNA extraction and PCR amplification
RNA extraction from HA positive allantoic fluid, was achieved using the Viral RNA mini kit (Qiagen, Germany), according to the manufacturer’s instructions. Reverse transcription was conducted using Uni12 primer: 5′-AGCAAAAGCAGG-3′. Reverse transcription (RT)-PCR was conducted using the One-Step RNA PCR Kit (AMV) (TaKaRa, China). All the segments were amplified with segment-specific primers as described elsewhere [17] (Table S1). The PCR products were purified with the Agarose Gel DNA Fragment Recovery Kit Ver.2.0 (TaKaRa, China), and the fragments were cloned into pGEM T easy vector (Promega).
Sequencing and phylogenetic analysis
The fragments sequencing were carried out using the Big Dye Terminator V.3.0 Cycle Sequencing Ready Reaction kit (ABI, Foster City, CA). The sequencing reactions were performed according to the manufacturer’s instructions. Sequencing of three independent clones of each PCR product was performed to eliminate errors resulting from the RT-PCR or cloning steps. The nucleotide sequences were deposited into GenBank under the accession numbers: JN605372–JN605403 and JN716319–JN716358. The sequences were analyzed by the BioEdit version 7.0.9.0 DNA analysis software [18]. Phylogenetic trees were constructed by MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0, applying the neighbor-joining method and Tamura–Nei model with bootstrap analysis (1,000 replicates) [19]. The reference sequences of strains used in this study were obtained from the Influenza Sequences Database (http://www.ncbi.nlm.nih.gov).
Results
Virus isolation and phylogenetic analysis
We have isolated 26 strains of AIVs, subtypes H1 (n = 9), H3 (n = 1), H4 (n = 5), H5 (n = 7), and H7 (n = 4) in markets in Zhejiang Province, China, in 2011. In this study, all the eight gene segments of the subtype H1 isolates were sequenced. The whole genomes of seven H1N3 AIVs, A/duck/Zhejiang/0611-8/2011(H1N3) (0611-8), A/duck/Zhejiang/0611-3/2011(H1N3) (0611-3), A/duck/Zhejiang/0611-15/2011(H1N3) (0611-15), A/duck/Zhejiang/0611-17/2011(H1N3) (0611-17), A/duck/Zhejiang/0611-19/2011(H1N3) (0611-19), A/duck/Zhejiang/0611-21/2011(H1N3) (0611-21), and A/duck/Zhejiang/0611-24/2011(H1N3) (0611-24) were identical, and they were compared to the nucleotide sequences available in the GenBank database, and they had the same sequence homology. So, we chose the isolate 0611-8 which represented the H1N3 AIVs. The sequence comparisons of H1 AIVs of the isolate 0611-8 and the other two H1N2 AIVs (A/duck/Zhejiang/0224-6/2011 and A/duck/Zhejiang/0607-13/2011) with their closest genetic relatives are shown in Table 1. Nucleotide sequences of H1N2 and H1N3 representing different geographic locations, host species, and different lineages were included for phylogenetic analysis (Supplementary material).
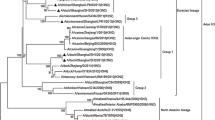
The results of phylogenetic analysis showed that all genes, HA, NA (N3), PB1, PB2, PA, NP, NA, M, and NS, of the H1 AIVs were clustered in the Eurasian lineage (Figs. 1, S1). All seven H1N3 AIVs, such as 0611-3, 0611-8, 0611-15, 0611-17, 0611-19, 0611-21, and 0611-24 strains, were identical, and they had a group in every gene phylogeny. The HA phylogenetic tree suggests that two different H1 genetic groups are co-circulating in Zhejiang Province. One group is formed by 0607-13, 0611-3, 0611-8, 0611-15, 0611-17, 0611-19, 0611-21, and 0611-24 strains, while the virus 0224-8 belongs to a distinct group. In the NA (N2) phylogenetic tree, the two H1N2 AIVs (0224-8 and 0607-13 strains) belong to Y439-like and Y280-like lineage, respectively.



Phylogenic analysis of HA and NA (N2 and N3) genes of the H1 influenza viruses isolate. The tree was created by the neighbor-joining method and bootstrapped with 1,000 replicates using the MEGA4.0 package. The H1 influenza viruses characterized are highlighted by a triangle. The scale bar represents the distance unit between sequence pairs
The HA gene of the virus 0607-13 (H1N2) shows the highest nucleotide similarity to A/wild duck/Korea/CW09/2005 (H1N1) at 97%. The NA gene of this virus has the highest nucleotide similarity to A/chicken/Jiande/01/2009 (H9N2) at 98% (Table 1). The HA, PB2, PB1, PA, NP, M, and NS gene phylogenies indicate that 0607-13, 0611-3, 0611-8, 0611-15, 0611-17, 0611-19, 0611-21, and 0611-24 had similar ancestors for these genes (Figs. 1, S1). According to the above-described analysis of phylogenetic relationships, the 0607-13 strain was a reassortant virus and derived its genes from different subtype from aquatic birds and poultry in Eastern Asia. This finding indicates that a re-assortment event between H1 and H9N2 occurred in ducks.
Molecular characterization
Based on the deduced amino acid sequences of the HA genes, the HA cleavage site pattern, IPSV(I)QSR/GL, of H1N2 and H1N3 isolates displayed features of a monobasic cleavage site. It is known that the addition of multiple amino acids at the cleavage site such as arginine (R) and lysine (K) may turn LPAI into HPAI in subtypes H5 and H7 AVIs [20].
Amino acids at receptor-binding sites, HA 98, 153, 155, 183, 190, 194, and 195 (H3 numbering system), were also analyzed (Table 2) [6, 21, 22]. In this study, the amino acids of the nine H1 AIVs at positions 224–229 and 134–138 were “RGQAGR” and “GVTAA,” respectively. The receptor-binding sites of H1N2 and H1N3 AIVs, Q226 and G228 were similar to all H1N2 and H1N3 AVIs, in both Eurasian and North American lineages suggesting that these viruses would preferentially bind to alpha 2–3 linked sialic acid receptors predominant in avian species [23, 24]. And there was no mutation of interest in the receptor-binding sites, which could increase affinity toward alpha 2–6 linked sialic acid receptors. However, whether these H1 AIVs possess low pathogenicity and have an ability to infect mammalian hosts requires further study.
In HA, seven potential glycosylation sites, sites 27, 28, 40, 104, 304, 498, and 557, were detected in the nine H1 AIVs. The specific polypeptide for N-linked glycosylation is defined as (Asn-X-Ser/Thr), where X can be any amino acid, except proline or aspartic acid [25].
In the NA of the 0607-13 (H1N2) virus, there was a deletion of three amino acids “TEI” (sites 63–65) at the NA stalk region, as previously described [26, 27]. It was revealed that the NA of 0607-13 (H1N2) virus shared a higher sequence similarity with H9N2 AIVs than other N2 AIVs.
NA inhibitors (oseltamivir and zanamivir) are effective antiviral drugs for the treatment and prophylaxis of influenza infections. The His275Tyr mutation is the molecular marker of oseltamivir resistance, and it was predicted previously that mutant viruses would be less viable than sensitive ones [28, 29]. The His275Tyr mutation was not observed in the NA of these nine H1 isolates.
It is notable that Val27Ala and Ser31Asn mutations in the M2 protein, which is associated with amantadine resistance of influenza virus [30], were not observed in the M2 of all the H1 AIV isolates.
Discussion
During 2000 to 2001, seven subtypes of LPAI, including H1N1, H2N9, H3N2, H3N3, H3N6, H4N6, and H9N2, from LPMs were identified in Central China [3]. The results of previous surveillance and the published data in GenBank revealed that H1 avian influenza viruses appeared in avian species in China [31]. To date, there were only three H1 AIV isolates, A/quail/Nanchang/12-340/2000 (H1N1), A/WDK/JX/12416/2005 (H1N1), and A/duck/Hebei/843/2005 (H1N2), isolated from avian species in China; and the information concerning the molecular characteristics of H1 AIVs was limited. This study highlights the first LPAI subtypes H1N3 AIVs ever reported in poultry from China. In this study, phylogenetic analysis showed that all eight genes of all H1 AIVs from Zhejiang Province belonged to the Eurasian lineage.
Genetic analysis has shown that the pandemic H1N1 2009 virus was originated from swine species through avian–human–swine “triple” re-assortment events [32]. The phylogenetic tree based on the HA gene (Fig. 1) showed the three lineages corresponding to avian, human, and swine influenza A viruses. It was revealed that the H1 AIVs were not the closest common ancestor of swine, human, and swine-origin human H1N1 viruses.
Table 1 suggests that PB2, NP, and NA genes of the virus 0607-13 (H1N2) had the highest nucleotide similarity to those of H5N1 avian-like swine influenza virus (A/swine/Fujian/F1/2001), H5N1 AIV (A/chicken/Shantou/2535/2001), and H9N2 AIV (A/chicken/Jiande/01/2009), respectively. A previous study showed that H9N2 AIVs isolated from poultry underwent extensive re-assortment to generate multiple novel genotypes in ducks and chickens, in Eastern China since 1998 [27]. It is possible that the 0607-13 virus has resulted from re-assortment of the H1N1, H5N1, and H9N2 viruses, or some LPAI viruses, including the 0607-13 virus, were the donors of the internal genes of avian H5N1 viruses in China.
Aquatic birds, including ducks, represent the major natural LPAI virus reservoir and have a higher prevalence of influenza A virus than other species [33–35]. Considering that the re-assorted H1N2 influenza viruses were created in ducks in LPMs, it would be possible that these ducks could play an important role in the re-assortment for creating H5N1 influenza viruses. In southern and eastern China, novel H9N2 influenza viruses that were double or even triple re-assortment were isolated in avian species [27, 36]. Some of them contained internal genes that are closely related to those in 1997 and 2001 H5N1 AIVs in Hong Kong [26, 36]. In 2006, HPAI H5N1 infection was reported in one patient who had visited LPMs in Southern China [37]. LPMs are considered a major source of influenza A virus dissemination (by direct or indirect contact) and potential influenza A virus re-assortment. In this study, although there was no evidence of re-assortment among the human, swine, and avian hosts, continued surveillance is needed considering the important role of the domestic duck in the dissemination and re-assortment of AIVs.
In conclusion, AIVs subtypes H1N2 and H1N3 were isolated from domestic ducks in LPMs in Zhejiang Province, Eastern China, in 2011. Whole genome sequences of the H1 AIVs were determined. Phylogenetic analysis of all the eight viral genes showed that the viruses clustered in the Eurasian lineage of influenza A viruses. The HA cleavage site of all the viruses displayed features of a monobasic cleavage site.
References
R.G. Webster, W.J. Bean, O.T. Gorman, T.M. Chambers, Y. Kawaoka, Microbiol. Rev. 56, 152–179 (1992)
Y. Kawaoka, T.M. Chambers, W.L. Sladen, R.G. Webster, Virology 163(1), 247–250 (1988)
M. Liu, S. He, D. Walker, N. Zhou, D.R. Perez, B. Mo, F. Li, X. Huang, R.G. Webster, R.J. Webby, Virology 305(2), 267–275 (2003)
C. Cardona, K. Yee, T. Carpenter, Poult. Sci. 88(4), 856–859 (2009)
D.C. Nguyen, T.M. Uyeki, S. Jadhao, T. Maines, M. Shaw, Y. Matsuoka, C. Smith, T. Rowe, X. Lu, H. Hall, X. Xu, A. Balish, A. Klimov, T.M. Tumpey, D.E. Swayne, L.P. Huynh, H.K. Nghiem, H.H. Nguyen, L.T. Hoang, N.J. Cox, J.M. Katz, J. Virol. 79(7), 4201–4212 (2005)
T. Wisedchanwet, M. Wongphatcharachai, S. Boonyapisitsopa, N. Bunpapong, P. Kitikoon, A. Amonsin, Virol. J. 8, 131 (2011)
H.J. Moon, M.S. Song, D.J. Cruz, K.J. Park, P.N. Pascua, J.H. Lee, Y.H. Baek, D.H. Choi, Y.K. Choi, C.J. Kim, Arch. Virol. 155(2), 229–241 (2010)
R.G. Webster, Lancet 363(9404), 234–236 (2004)
M. Peiris, K.Y. Yuen, C.W. Leung, K.H. Chan, P.L. Ip, R.W. Lai, W.K. Orr, K.F. Shortridge, Lancet 354, 916–917 (1999)
Y.P. Lin, M. Shaw, V. Gregory, K. Cameron, W. Lim, A. Klimov, K. Subbarao, Y. Guan, S. Krauss, K. Shortridge, R. Webster, N. Cox, A. Hay, Proc. Natl Acad. Sci. USA 97, 9654–9658 (2000)
K.R. Cameron, V. Gregory, J. Banks, I.H. Brown, D.J. Alexander, A.J. Hay, Y.P. Lin, Virology 278(1), 36–41 (2000)
W.M. Jiang, S. Liu, J. Chen, G.Y. Hou, J.P. Li, Y.F. Cao, Q.Y. Zhuang, Y. Li, B.X. Huang, J.M. Chen, J. Gen. Virol. 91(Pt 10), 2491–2496 (2010)
K. Ji, W.M. Jiang, S. Liu, J.M. Chen, J. Chen, G.Y. Hou, J.P. Li, B.X. Huang, J. Virol. Methods 163(2), 186–189 (2010)
B.S. Kim, H.M. Kang, J.G. Choi, M.C. Kim, H.R. Kim, M.R. Paek, J.H. Kwon, Y.J. Lee, Poult. Sci. 90(7), 1449–1461 (2011)
J. Chen, Z. Yang, Q. Chen, X. Liu, F. Fang, H. Chang, D. Li, Z. Chen, Virus Genes 38(1), 66–73 (2009)
Office International des Epizootics. Manual of Standards for Diagnostic Tests and Vaccines, 3rd edn. (OIE, Paris, 1996), pp. 155–160
E. Hoffmann, J. Stech, Y. Guan, R.G. Webster, D.R. Perez, Arch. Virol. 146(12), 2275–2289 (2001)
N.Z. Jandaghi, T.M. Azad, M. Naseri, J. Yavarian, R. Nategh, Arch. Virol. 155(5), 717–721 (2010)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24(8), 1596–1599 (2007)
T. Horimoto, T. Ito, D.J. Alexander, Y. Kawaoka, J. Vet. Med. Sci. 57(5), 927–930 (1995)
S.J. Gamblin, L.F. Haire, R.J. Russell, D.J. Stevens, B. Xiao, Y. Ha, N. Vasisht, D.A. Steinhauer, R.S. Daniels, A. Elliot, D.C. Wiley, J.J. Skehel, Science 303(5665), 1838–1842 (2004)
J. Stevens, A.L. Corper, C.F. Basler, J.K. Taubenberger, P. Palese, I.A. Wilson, Science 303(5665), 1866–1870 (2004)
J. Liu, D.J. Stevens, L.F. Haire, P.A. Walker, P.J. Coombs, R.J. Russell, S.J. Gamblin, J.J. Skehel, Proc. Natl Acad. Sci. USA 106(40), 17175–17180 (2009)
A.C. Bateman, M.G. Busch, A.I. Karasin, N. Bovin, C.W. Olsen, J. Virol. 82, 8204–8209 (2008)
A. Helenius, M. Aebi, Annu. Rev. Biochem. 73, 1019–1049 (2004)
Y. Guan, K.F. Shortridge, S. Krauss, R.G. Webster, Proc. Natl Acad. Sci. USA 96, 9363–9367 (1999)
P. Zhang, Y. Tang, X. Liu, W. Liu, X. Zhang, H. Liu, D. Peng, S. Gao, Y. Wu, L. Zhang, S. Lu, X. Liu, J. Virol. 83(17), 8428–8438 (2009)
F.Y. Aoki, G. Boivin, N. Roberts, Antivir. Ther. 12, 603–616 (2007)
H. Zaraket, R. Saito, Y. Suzuki, T. Baranovich, C. Dapat, I. Caperig-Dapat, H. Suzuki, J. Clin. Microbiol. 48, 1085–1092 (2010)
V.M. Deyde, X. Xu, R.A. Bright, M. Shaw, C.B. Smith, Y. Zhang, Y. Shu, L.V. Gubareva, N.J. Cox, A.I. Klimov, J. Infect. Dis. 196(2), 249–257 (2007)
J. Liu, Y. Bi, K. Qin, G. Fu, J. Yang, J. Peng, G. Ma, Q. Liu, J. Pu, F. Tian, J. Clin. Microbiol. 47(8), 2643–2646 (2009)
G. Neumann, T. Noda, Y. Kawaoka, Nature 459, 931–939 (2009)
B. Olsen, V.J. Munster, A. Wallensten, J. Waldenström, A.D. Osterhaus, R.A. Fouchier, Science 312(5772), 384–388 (2006)
O.M. Jeong, Y.J. Kim, J.G. Choi, H.M. Kang, M.C. Kim, J.H. Kwon, Y.J. Lee, Virus Genes 42(1), 55–63 (2011)
S.J. Kang, H.M. Kim, Y.H. Kim, S.D. Hwang, J.S. Shin, K.B. Ku, H.S. Kim, S.H. Seo, Virus Genes 38(1), 80–84 (2009)
K.S. Li, K.M. Xu, J.S. Peiris, L.L. Poon, K.Z. Yu, K.Y. Yuen, K.F. Shortridge, R.G. Webster, Y. Guan, J. Virol. 77, 6988–6994 (2003)
M. Wang, B. Di, D.H. Zhou, B.J. Zheng, H. Jing, Y.P. Lin, Y.F. Liu, X.W. Wu, P.Z. Qin, Y.L. Wang, L.Y. Jian, X.Z. Li, J.X. Xu, E.J. Lu, T.G. Li, JXu Emerg, Infect. Dis. 12(11), 1773–1775 (2006)
Acknowledgments
This study was supported by Grants from the National Science Foundation of the People’s Republic of China (30872163), National Key Technologies R&D Programme for the 12th Five-Year Plan of China (2012ZX1000-004-005) and the State Key Laboratory of independent task (No. 2010ZZ04).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Wu, Hb., Guo, Ct., Lu, Rf. et al. Genetic characterization of subtype H1 avian influenza viruses isolated from live poultry markets in Zhejiang Province, China, in 2011. Virus Genes 44, 441–449 (2012). https://doi.org/10.1007/s11262-012-0716-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0716-y