
Abstract
An outbreak of echovirus 30 (E-30) in 2009 was confirmed by both frequent isolation of the virus from sewage as well as from patient samples in Finland. Over the last 10 years E-30 had only been isolated sporadically in Finland. We here study the phylogenetic relationships of the strains from the outbreak in the context of E-30 circulation over the last 20 years. The analyzed region comprised 276 nucleotides in the 5′ end of VP1 (nucleotides 132–407 in the VP1 of the E-30 Bastianni strain). The Finnish strains were clustered into at least four distinct genogroups, with seven clusters exceeding the genotype demarcation of 12% and the 2009 epidemic strains forming the largest genogroup VII. Moreover, we detected largely divergent genotypes in 2007 and 2009. Interestingly, close genetic relatives of the epidemic strains had already been isolated a few years before the outbreak. Phylodynamic analysis estimated 8.9 years (95% highest posterior density intervals 7.0–11.0) as the age of genogroup VII, indicating a probable origin and evolutionary history prior to its introduction and epidemic expansion in Finland. Finally, the most recent common ancestor for the current E-30 diversity dates back to 1939 (95% highest posterior density intervals 1913–1956).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Echovirus 30 (E-30) is one of the enteroviruses recurring in reports on meningitis outbreaks and circulating in human communities. It is often associated with more serious infections that lead to neurological symptoms and hospital care. However, enterovirus infections involving E-30 are primarily mild or sub-clinical, indicating that the diagnostically confirmed cases with serious symptoms represent only the tip of the iceberg.
Taxonomically E-30 belongs to Human enterovirus B (HEV-B) species in the genus Enterovirus, family Picornaviridae [1]. Picornaviruses are small, non-enveloped virus particles with capsids characterized by icosahedral symmetry. Their genome is an approximately 7,500 nt long messenger-sense RNA with one open reading frame coding for capsid proteins VP1-4 and seven non-structural proteins. Their evolutionary dynamics are fueled by a rapid accumulation of point mutations due to the lack of proof-reading and correction mechanisms of the RNA-dependent RNA polymerase and frequent recombination. These processes enable the virus populations to readily adapt to changing environmental conditions and efficiently colonize a niche for propagation.
At the turn of the millennium, we reported increased epidemic activity of E-30 in Finland [2]. In tandem several other reports described similar findings, indicating a global predominance of a single epidemic genotype [3–5]. These reports were followed by global descriptions of E-30 outbreaks [6–15]. Long-term surveillance of E-30 reporting by the Centers for Disease Control and Prevention has revealed the periodicity of E-30 outbreaks within a community [10].
In Finland most years will see a few E-30 strains genetically typed. Typically, detection is in environmental samples of sewage water. However, in 2009 the epidemic activity of E-30 dramatically increased after years of sporadic findings. E-30 was readily detected in sewage from different parts of the country while specimens from patients with neurological symptoms also began to accumulate. Here, we present a phylogenetic and phylodynamic analysis of the strains that made up the recent Finnish outbreak.
Materials and methods
Viruses
The Echovirus 30 strains analyzed in this study originate from environmental surveillance of enteroviruses in Finland, from clinical specimens studied to determine the etiology of viral meningitis, from research projects [16], and from clinical HEV isolates sent to the national enterovirus reference laboratory from other Finnish clinical virology laboratories [17]. All strains where a partial VP1 sequence existed were included in the study. In sum 108 strains were available, of which 79.6% originated from environmental samples and 20.3% from clinical specimens, e.g., stool and cerebrospinal fluid (Tables 1, 2). A total of 27 strains were isolated during the period 1993–2008, while the other 81 strains originated from the 2009 outbreak. The 2009 outbreak strains were isolated from different cities in Finland (Table 2). The cell lines used for isolation included human rhabdomyosarcoma, human colorectal adenocarcinoma, human cervical carcinoma, and the green monkey kidney cell line (GMK). In addition, VP1 sequences from [2] were included in the analysis. Altogether, those international strains were collected over a time span of 35 years (1975–2009), while isolates from Finland covered a 20-year period (1985–2009). The GenBank accession numbers of the sequences reported in this paper are: HM366609-HM366716, and the alignment is available on request from the authors.
Isolation of RNA, RT-PCR, sequencing, and molecular typing
RNA was extracted from 100 µl of the infected cell cultures by commercial procedures (E.Z.N.A.® Total RNA kit, Omega Bio-Tek Inc., Doraville, GA, USA) according to the manufacturer’s instructions. RT-PCR was performed with primers 292 and 222 [18, 19], designed for identification of all enteroviruses. The purification of PCR products, sequencing, and molecular typing were done as described in Blomqvist et al. [17].
Phylogenetic analysis
Multiple sequence alignments were made with MEGA 4.0 [20] using default parameters and thereafter manually optimized according to the amino acid translation. The phylogenetic tree was estimated with MEGA 4.0 using Neighbor-joining [21] with the maximum composite likelihood model [22]. The transition/transversion ratio was estimated from the data set (4.2). Bootstrapping was performed using 1,000 pseudo-replicates [23].
Phylodynamic analysis
Rate of evolution and divergence times were estimated from the partial VP1 sequences using a Bayesian Monte Carlo Markov Chain (MCMC) method and implemented in BEAST version 1.5.1 [24]. Bayesian MCMC analyses were performed using a relaxed molecular clock model (the uncorrelated log-normal distributed model) [25]. Bayesian analysis was run for 100 million states and sampled every 100,000 states. Posterior probabilities were calculated with a burn-in of 1 million states and checked for convergence using Tracer version 1.4.1 [26].
Results
In 2009 the epidemic activity of E-30 dramatically increased in Finland (Fig. 1). After years of low detection, E-30 was isolated from sewage as well as from patient samples from all over the country. Samples from only a few representative cases/epidemic were collected and analyzed. All of the patients from whom the anamnesis data were available suffered from neurological complaints: headache, nausea, and nuchal rigidity being the most common symptoms. Most of the patients were diagnosed with meningitis.

Echovirus 30 detections in THL/Finland during 1993–2009. Data from 1985 to 1992 were obtained from Savolainen et al. [2]
The seasonal distribution of total E-30 detections in Finland from 1985 to 2009 showed a curve typical of other HEVs, peaking in the autumn (Fig. 2). Yet, sporadic detections occurred throughout the year. Also the 2009 outbreak occurred at the time of the enterovirus high season, in autumn (Table 2), although a genetically almost identical strain had been isolated already in March, 2009 (HM366621, Fig. 3).

Monthly distribution of echovirus 30 detections in Finland. All detections are included for which isolation date data were available. Data are collected from the period 1985 to 2009. Data from 1985 to 1992 were obtained from Savolainen et al. [2]

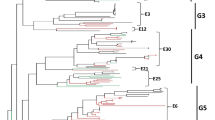
Evolutionary relationships of the Finnish E-30 strains and reference strains in the 276 nucleotide region in the 5′-part of VP1. The evolutionary history was inferred using the Neighbor-joining method. Evolutionary distances were computed using the Maximum Composite Likelihood model. The strains of this study are shown in black while reference sequences from GenBank are shown in red. Sequences from Savolainen et al. [2] are shown in blue. Numbers represent bootstrap values for each branch calculated with 1,000 replicates. Colored ovals indicate genogroups represented by the Finnish strains of this study. Black circles indicate strains of genotype VII originating prior to 2009. The estimated year of emergence of the lineages are shown below genogroup denotations with 95% highest posterior density intervals. The rectangular tree in the lower left corner represents clustering with echovirus 21 (E21)-Farina (AY302547) as an outgroup (Color figure online)
A total of 112 Finnish strains were used in the phylogenetic analysis together with 63 strains from [2] and the E-30 prototype strain Bastianni (Fig. 3). In addition, based on prior BLAST searches in GenBank (February, 2010), 19 previously published sequences were included. The analyzed region comprised 276 nucleotides in the 5′ end of VP1 (nucleotides 132–407 in the VP1 of the E-30 Bastianni strain). The Finnish strains clustered into at least four distinct genetic lineages or genotypes with high bootstrap support. Based on a demarcation of 12% of genetic divergence [2] seven lineages could be identified. Bootstrap support values for genogroups IV–VI remained low, but taken the evident clustering, these lineages were considered as genotypes in the subsequent analyses (I–VII, Fig. 3). One strain (HM366709, Fig. 3) did not cluster with any other strains. Genogroup V comprised strains from the largest time span, from 2000 to 2008, while all other lineages were temporally more restricted. The largest genogroup, VII, comprised most strains from 2009 and only a few strains from other years (marked with a black dot in Fig. 3). The closest GenBank matches to the 2009 outbreak strains were found to have originated in France and the United Kingdom. Two recent Finnish strains that diverged most from all other strains were shown to have their closest relatives in the designated ancient genogroups, previously isolated from Columbia and the Russian states [4, 13] (HM366613 and HM366623, genogroup I, Fig. 3).
The maximum nucleotide distance between E-30 strains in this study in the analyzed genomic region was 23.6%. The maximum amino acid distance between all strains was 11.0%. The ranges of distance values for sequence relationships within genogroups are shown in Table 3. The sequence data originated from genetic typing of HEVs and thus did not include the complete VP1, but was a part of it. This reduction may have an effect on the topology and also on the robustness of the bootstrapping analysis. Therefore, not all observed genogroups had a significant bootstrap support.
The phylodynamic analysis of the partial VP1 sequences suggested that the mean evolutionary rate of the E-30 lineages was 6.2 × 10−3 (95% highest posterior density intervals (HPD) 4.7–7.8 × 10−3) substitutions/site/year. The standard deviation of the lognormal relaxed clock was fairly high with a coefficient of variation of 1.07 (95% HPD 0.73–1.40), meaning that rates were varying across lineages within about 100% of the mean rate. Assuming this relaxed estimated molecular clock, the age of E-30’s tree root was 71.1 (52.9–95.4) years for those strains included in this analysis and the age of group VII (epidemic strains from 2009) was 8.9 (7.0–11.0) years. The estimated times of emergence for each lineage are shown in Fig. 3.
Discussion
Echovirus 30 in Finland
This article analyzes E-30 isolates collected in Finland during the period 1985–2009 (Fig. 1). From 1996 to 1997 there was an outbreak of E-30 in Finland that manifested through both clinical and environmental virus detections (Fig. 1). We have previously reported a molecular epidemiological analysis of the E-30 outbreak strains and their relation to global genotypic variation [2]. Thereafter, E-30 was occasionally detected in sewage surveillance samples from 1998 to 2006. In 2007, a slight increase in the environmental detections of E-30 was found that continued into 2008. During these 2 years E-30 was also detected in clinical cases after an absence of 10 years. This was a prelude to the E-30 outbreak in 2009, when E-30 was readily detected in sewage around Finland as well as from patient samples (Figs. 1, 3).
Potential factors leading to the 2009 echovirus 30 outbreak in Finland
Close genetic relatives of the 2009 epidemic virus lineage were detected already in 2003 and again in 2007–2008. A likely source for the lineage is importation from other European countries. Genetically similar strains were isolated during outbreaks in France in 2005 [27] and 2007 [28] and in the United Kingdom in 2007 and 2008 [29]. Moreover, the results of the divergence analysis support importation as the source of the strains, as the estimated mean age of the lineage, 8.9 years (7.0–11.0), outdates the oldest Finnish detection of this lineage’s strains from 2003. However, one may also speculate that the earlier solitary members of the lineage may have been missed in the surveillance.
One can only speculate on the factors that caused the originally silent circulation to expand to an outbreak. It has been suggested that a critical mass of infants or other unprotected members of the community has to be reached before a viral epidemic can emerge. However, in this outbreak most of the specimens from hospitalized patients were from teenagers (mean/median age of 16.1/15.0 years) who were also unprotected 2 years earlier. A similar finding has previously been reported for the US; E-30 is most commonly detected in older children and adults [10]. It has been shown that outbreaks are usually caused by a new genomic lineage [2, 4, 5]. It is possible that the virus has drifted antigenically to produce a variant with greater capacity to infect susceptible hosts that were immune to earlier variants. We have previously shown that antigenic differences between the genetically diverse E-30 strains existed; however, their association to genetic clustering was not straightforward, most probably because antigenic epitopes are not restricted to VP1 [30]. In the case of the current outbreak, however, the low amino acid sequence variability between the earlier and epidemic strains makes this explanation less plausible, as discussed previously [31]. Also, our previous studies indicate that E-30 strains have strain-specific characteristics that determine their ability to destroy human pancreatic islet β-cells [32]. These characteristics were associated with genome properties found outside the region of structural protein-coding [30].
Studies of the underlying evolutionary basis for these characteristics have revealed recombination in the non-structural part of the genome as a major factor in the molecular evolution of E-30 [31, 33, 34]. It has also been hypothesized that frequent recombination, possibly with another HEV-B virus, would provide an E-30 strain with unusual properties [13] and possibly enhance transmissibility [31]. The lack of evidence for recombination in the evolutionary history of the current E-30 strains obviously reduces any discussion on the effect of recombination to a hypothetical level. In 2007, a divergent E-30 strain was detected in Kotka (HM366613) and in 2009 in Helsinki (HM366623) (Fig. 3). These strains likely represent importation from Russia and its neighboring states, as the closest sequences available in GenBank originated from Finland’s eastern neighbor. Hypothetically, while co-circulating with these more divergent lineages [13], a predecessor of the epidemic strain may have received genetic material from them that promoted transmissibility, leading to a nationwide epidemic. Virus migration has previously been considered a major factor in E-30 diversity and the occurrence of aseptic meningitis [28]. However, extensive complete genome sequence analyses are needed to resolve the role of recombination in the formation of the Finnish epidemic virus, let alone identifying the underlying genetic and/or phenotypic determinants.
Phylodynamic observations
The estimated mean rate of molecular evolution, 6.2 × 10−3 (4.7–7.8 × 10−3) substitutions/site/year, based on the phylodynamic analysis is congruent with rates estimated for E-30 and other HEV-B viruses, although sequences of only 276 nt were used for the current analysis. For the partial VP1 gene, the E-30 rate has been estimated to be 8.5 × 10−3 (7.0–9.9 × 10−3) substitutions/site/year [31]; for enterovirus 71 (EV-71) the rate was recently estimated at 4.2–4.5 × 10−3 (4.0–4.8 × 10−3) substitutions/site/year [35]. HEV-B viruses seem to evolve more slowly than polioviruses, for which the rate has been estimated to be 1.0–2.6 × 10−2 substitutions/site/year [36, 37]. The estimated year for the root of the tree, 1939 (95% HPD 1913–1956), is also concordant with other HEV-B virus diversification time estimates, although inclusion of older strains places it a little further back in time, as estimated for another data set of E-30 [31]. The origin of EV-71 has been estimated to have occurred in 1941 (95% CI 1929–1952) [35], while the coxsackievirus B5 (CV-B5) ancestor of the bifurcating lineages was dated to 1854 (95% HPD 1807–1898) [38]. Our estimate of the root would place the evolutionary age of E-30 lineages in between CV-B5 and EV-71.
Environmental surveillance
The rise in the E-30 detections from sewage can already be noticed before the mass of clinical cases initiated in 2009 (Fig. 1). Environmental surveillance of sewage water is an excellent method for monitoring the circulation of enteric viruses in populations. The detected clinical isolates of enteroviruses only represent the most severe infections and are indicative of an extensive circulation of viruses resulting in mild or asymptomatic infections. Environmental surveillance is increasingly exploited in the surveillance of wild and vaccine-derived poliovirus circulation [39]. It reveals silent circulation in the communities and can help to indicate onsets of outbreaks. Thus, environmental surveillance can be a valuable tool in timing measures to control cases of severe infection that would have potentially crippling effects on communities.
Conclusions
The 2009 E-30 outbreak was caused by viruses whose close genetic relatives had already been circulating in Finland in the years preceding. Potentially, recombination with co-circulating divergent lineages may have empowered the pre-epidemic strains to cause a nationwide outbreak.
References
G. Stanway, F. Brown, P. Christian, T. Hovi, T. Hyypiä, A.M.Q. King, N.J. Knowles, S.M. Lemon, P.D. Minor, M.A. Pallansch, A.C. Palmenberg, T. Skern, in Virus Taxonomy Eighth Report of the International Committee on Taxonomy of Viruses, ed. by C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger, L.A. Ball (Elsevier/Academic Press, London, 2005), pp. 757–778
C. Savolainen, T. Hovi, M.N. Mulders, Arch. Virol. 146, 521–537 (2001)
Centers for Disease Control and Prevention, MMWR (2003), pp. 761–783
M.S. Oberste, K. Maher, M.L. Kennett, J.J. Campbell, M.S. Carpenter, D. Schnurr, M.A. Pallansch, J. Clin. Microbiol. 37, 3928–3933 (1999)
G. Palacios, I. Casas, D. Cisterna, G. Trallero, A. Tenorio, C. Freire, J. Virol. 76, 4940–4949 (2002)
C.M. Castro, D.S. Oliveira, O. Macedo, M.J. Lima, M.B. Santana, A.L. Wanzeller, E. Silveira, L. Gomes Mde, Mem. Inst. Oswaldo Cruz. 104, 444–450 (2009)
E.M. Begier, M.S. Oberste, M.L. Landry, T. Brennan, D. Mlynarski, P.A. Mshar, K. Frenette, T. Rabatsky-Ehr, K. Purviance, A. Nepaul, W.A. Nix, M.A. Pallansch, D. Ferguson, M.L. Cartter, J.L. Hadler, Clin. Infect. Dis. 47, 616–623 (2008)
B. Kapusinszky, K.N. Szomor, A. Farkas, M. Takacs, G. Berencsi, Virus Genes 40, 163–173 (2010)
Y.N. Zhao, Q.W. Jiang, R.J. Jiang, L. Chen, D.S. Perlin, Emerg. Infect. Dis. 11, 562–567 (2005)
N. Khetsuriani, A. Lamonte-Fowlkes, S. Oberst, M.A. Pallansch, MMWR Surveill. Summ. 55, 1–20 (2006)
T.V. Amvrosieva, N.V. Paklonskaya, A.A. Biazruchka, O.N. Kazinetz, Z.F. Bohush, E.G. Fisenko, Cent. Eur. J. Public Health 14, 67–73 (2006)
D. Brunel, N. Leveque, J. Jacques, F. Renois, J. Motte, L. Andreoletti, J. Clin. Virol. 42, 225–228 (2008)
A.N. Lukashev, O.E. Ivanova, T.P. Eremeeva, L.V. Gmyl, J. Clin. Microbiol. 46, 665–670 (2008)
T. Hayashi, T. Shirayoshi, T. Nagano, H. Yaoita, S. Kogure, H. Nariai, T. Natsumeda, M. Taniuchi, M. Sandoh, Y. Sato, Intern. Med. 48, 1767–1771 (2009)
G. Trallero, A. Avellon, A. Otero, T. De Miguel, C. Perez, N. Rabella, G. Rubio, J.E. Echevarria, M. Cabrerizo, J. Clin. Virol. 47, 170–176 (2010)
P. Klemola, S. Kaijalainen, P. Ylipaasto, M. Roivainen, Ann N Y Acad Sci 1150, 210–212 (2008)
S. Blomqvist, A. Paananen, C. Savolainen-Kopra, T. Hovi, M. Roivainen, J. Clin. Microbiol. 46, 2410–2413 (2008)
M.S. Oberste, K. Maher, M.R. Flemister, G. Marchetti, D.R. Kilpatrick, M.A. Pallansch, J. Clin. Microbiol. 38(3), 1170–1174 (2000)
M.S. Oberste, W.A. Nix, K. Maher, M.A. Pallansch, J. Clin. Virol. 26(3), 375–377 (2003)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 2007, 1596–1599 (2007)
N. Saitou, M. Nei, Mol. Biol. Evol. 4, 406–425 (1987)
K. Tamura, M. Nei, S. Kumar, Proc. Natl Acad. Sci. USA 101, 11030–11035 (2004)
D.M. Hillis, J.J. Bull, Syst. Biol. 42, 182–192 (1993)
A.J. Drummond, A. Rambaut, BMC Evol. Biol. 7, 214 (2007)
A.J. Drummond, S.Y. Ho, M.J. Phillips, A. Rambaut, PLoS Biol. 4, e88 (2006)
A. Rambaut, A.J. Drummond, Tracer v1.4. Available from http://beast.bio.ed.ac.uk/Tracer (2007)
N. Leveque, J. Jacques, F. Renois, D. Antona, M. Abely, J.J. Chomel, L. Andreoletti, J. Clin. Virol. 48, 137–141 (2010)
J.L. Bailly, A. Mirand, C. Henquell, C. Archimbaud, M. Chambon, F. Charbonne, O. Traore, H. Peigue-Lafeuille, Infect. Genet. Evol. 9, 699–708 (2009)
E.C. Leitch, H. Harvala, I. Robertson, I. Ubillos, K. Templeton, P. Simmonds, J. Clin. Virol. 44, 119–124 (2009)
A. Paananen, C. Savolainen-Kopra, S. Kaijalainen, O. Vaarala, T. Hovi, M. Roivainen, J. Med. Virol. 79, 945–955 (2007)
E.C. McWilliam Leitch, J. Bendig, M. Cabrerizo, J. Cardosa, T. Hyypia, O.E. Ivanova, A. Kelly, A.C. Kroes, A. Lukashev, A. MacAdam, P. McMinn, M. Roivainen, G. Trallero, D.J. Evans, P. Simmonds, J. Virol. 83, 2109–2118 (2009)
M. Roivainen, P. Ylipaasto, C. Savolainen, J. Galama, T. Hovi, T. Otonkoski, Diabetologia 45, 693–702 (2002)
A.N. Lukashev, V.A. Lashkevich, O.E. Ivanova, G.A. Koroleva, A.E. Hinkkanen, J. Ilonen, J. Gen. Virol. 86, 3281–3290 (2005)
A. Mirand, C. Henquell, C. Archimbaud, H. Peigue-Lafeuille, J.L. Bailly, J. Gen. Virol. 88, 166–176 (2007)
K.K. Tee, T.T. Lam, Y.F. Chan, J.M. Bible, A. Kamarulzaman, C.Y. Tong, Y. Takebe, O.G. Pybus, J. Virol. 84, 3339–3350 (2010)
J. Jorba, R. Campagnoli, L. De, O. Kew, J. Virol. 82, 4429–4440 (2008)
G.V. Gavrilin, E.A. Cherkasova, G.Y. Lipskaya, O.M. Kew, V.I. Agol, J. Virol. 74, 7381–7390 (2000)
M. Gullberg, C. Tolf, N. Jonsson, M.N. Mulders, C. Savolainen-Kopra, T. Hovi, M. Van Ranst, P. Lemey, S. Hafenstein, A.M. Lindberg, J. Virol. 84(19), 9695–9708 (2010)
T. Hovi, Biologicals 34, 123–126 (2006)
Acknowledgments
The study was supported by grants from the Academy of Finland, Juvenile Diabetes Research Foundation and Päivikki and Sakari Sohlberg Foundation. Most of the isolated echovirus strains were derived from poliovirus surveillance projects supported by WHO (TSA 18/181/526). Dr Eeva Ruotsalainen, Helsinki University Central Hospital, is thanked for sending specimens. Mervi Eskelinen, Päivi Hirttiö, Alena Kaijalainen, Elisa Lamminsalo, Marja-Liisa Ollonen, and Eija Penttilä are acknowledged for excellent technical assistance. PL is supported by a postdoctoral fellowship from the Fund for Scientific Research (FWO), Flanders.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Savolainen-Kopra, C., Paananen, A., Blomqvist, S. et al. A large Finnish echovirus 30 outbreak was preceded by silent circulation of the same genotype. Virus Genes 42, 28–36 (2011). https://doi.org/10.1007/s11262-010-0536-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-010-0536-x