
Abstract
Echoviruses belong to the genus Enterovirus in the Picornaviridae family, forming a large group of Enterovirus B (EV-B) within the Enteroviruses. Previously, Echoviruses were classified based on the coding sequence of VP1. In this study, we performed a reliable phylogenetic classification of 277 sequences isolated from 1992 to 2019 based on the full-length genomes of Echovirus. In this report, phylogenetic, phylogeographic, recombination, and amino acid variability landscape analyses were performed to reveal the evolutional characteristics of Echovirus worldwide. Echoviruses were clustered into nine major clades, e.g., G1–G9. Phylogeographic analysis showed that branches G2–G9 were linked to common strains, while the branch G1 was only linked to G5. In contrast, strains E12, E14, and E16 clustered separately from their G3 and G7 clades respectively, and became a separate branch. In addition, we identified a total of 93 recombination events, where most of the events occurred within the VP1-VP4 coding regions. Analysis of amino acid variation showed high variability in the a positions of VP2, VP1, and VP3. This study updates the phylogenetic and phylogeographic information of Echovirus and indicates that extensive recombination and significant amino acid variation in the capsid proteins drove the emergence of new strains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Echoviruses belong to the largest sub-group of Enteroviruses (EVs), which is a member of the Enterovirus genus in the Picornaviridae family. Human enteroviruses are widely prevalent in the world, consisting of A, B, C, and D species (Brouwer et al., 2021). Echovirus is grouped in the Enterovirus B (EV-B) and is the most frequently detected species worldwide (Brouwer et al., 2021). Echovirus can infect multiple organs of human body with a wide variety of manifestations from acute or asymptomatic febrile illness of young children and infants to fatal aplastic anemia, encephalitis, and pulmonary hypertension (Rao, 2015). Different serotypes lead to different clinical signs. For example, E4 (Handsher et al., 1999), E6 (Fratty et al., 2023), E9 (Zhu et al., 2016), E13 (Diedrich & Schreier, 2001), E18 (Jiang et al., 2022), E30 (Castro et al., 2009), E33 (Huang et al., 2003) infections cause neurological disorders; E3 (Miyata et al., 2014), E11 (Hirade et al., 2023), and E18 (Xi et al., 2023) are associated with neonatal systemic illness; E21 is associated with hepatitis (Pedrosa et al., 2013). In addition, the cases of hand, foot, and mouth disease (HFMD) has been reported to be associated with E7 infection in China Mainland (Yao et al., 2015).
Echoviruses are small envelope-free, single-stranded positive-sense RNA viruses. The genome of Echovirus is approximately 7400 nucleotides long and contains a long open reading frame (ORF) flanked by 5’- and 3’-untranslated regions (UTRs). The ORF is translated into a polyprotein, which is then cleaved into precursor proteins P1, P2, and P3. P1 is further cleaved into structural proteins VP1-VP4. P2 and P3 are processed into six non-structural proteins including 2A, 2B, 2C (RNA helicase), 3A, C3 peptidase, and RNA-dependent RNA polymerase (RdRp) (Paul & Wimmer, 2015). VP1-VP4 proteins are major structural capsid proteins. Since the VP1 contains important antigen sites, it has high specificity for different serotypes. Therefore, the serotypes are widely classified according to VP1 coding sequence (Xiao et al., 2020).
The first discovery of Echovirus was in 1951, when it was isolated from stool samples of asymptomatic individuals (Ramos-Alvarez & Sabin, 1954). Currently, 28 different potential serotypes of Echoviruses have been reported, including E1-E7, E9, E11-E21, E24-E27 and E29-E33, whereas types E8, E10, E22, E23, E28 and E34 have been classified as other viruses (Home of Picornaviruses, 2023; Zhang et al., 2022). Echovirus has been found in many countries of Asia, Europe, Oceania, Africa, and Americas, including China (Zhang et al., 2022), India (Lavania et al., 2022), Switzerland (Grädel et al., 2022), France (Morsli et al., 2021), New Zealand (Dilcher et al., 2022), Australia (Roberts et al., 2020), Nigeria (Faleye et al., 2019), Brazil, and other countries (Sousa et al., 2019). Up to present, there are no approved vaccines or effective antiviral drugs available for the treatment of Echovirus infection (Chen et al., 2022; Ogbole et al., 2018).
Based on VP1 coding sequences, Echovirus isolates from different regions are classified into different sub-groups by different studies. For example, E1 strains collected in the Buenos Aires Metropolitan Area and Argentina during 1951–2016 were classified into two monophyletic subgroups, cluster I and II (Lizasoain et al., 2021). E3 strains isolated in Larissa, Thessaly, and Greece in 2009 were divided into three major monophyletic genogroups A–C, which were further subdivided into seven sub-genogroups (A1–A3, B1 and B2, C1 and C2) (Kyriakopoulou et al., 2015a). E6 strains identified in Poland were divided into four different major groups (I–IV) (Wieczorek et al., 2017). From 1953 to 2017, ninety-four representative strains of E11 from China and around the world were classified into six different genotypes (A–F). Genotypes A, C and D could be further segmented into A1–A5, C1–C4 and D1–D5 (Li et al., 2019; Oberste et al., 2003).
Previously, the Echovirus was divided into two clades (clade I and II) by phylogenetic analysis of VP1 coding nucleotide sequences isolated from patients diagnosed in Zhejiang, China during 2014–2017, with clade I including E9, E11, E14, E16 and E18, clade II including E6, E21, E25, E30 and E33 (Sun et al., 2019). However, with the increasing appearance of genetic variation and recombination among Echovirus strains, this may not be enough to explain the genetic diversity of these viruses. We have now thoroughly explored this issue by phylogenetic and phylogeographic analyses of the full-length virus genomes identified world-wide.
Echovirus classification is extremely complex, which needs an updated, robust, and well-categorized system to better understand the Echovirus infections. Thus, we accessed the NCBI GenBank database in this study to retrieve the full-length genome sequences of 277 Echoviruses isolated from 1992 to 2019, analyzed the phylogenetic correlations between them and mapped the phylogeographic network of the viruses. The results of this study can better clarify the previous classification, evolutionary features and phylogeographic distribution of Echovirus strains and provide useful hints on the genetic basis of their emergence and re-emergence.
Materials and Methods
Dataset
The available 277 full-length genome sequences of the Echovirus were retrieved from NCBI GenBank. The viruses were isolated during 1992 - 2019 from twenty-six different countries, where the highest number of sequences were reported from China (99), followed by the USA (43), Israel (25), Japan (15), Finland (12) and Spain (12) et al. (Table 1). In addition, the amino acid sequences of 277 full-length Echovirus polyproteins were also retrieved from NCBI GenBank. The viruses were identified by their GenBank ID, Virus name, country/region, and year of report, e.g., ID-virus name-country/region-year.
Construction of Phylogenetic Tree and Similarity Analysis
All the nucleotide or amino acid sequences were aligned with the ClustalW multiple sequence alignment tool in the MEGA11 software (Pennsylvania State University) (Tamura et al., 2021), in which all identical sequences or sequences containing large missing part were discarded. The sequences were manually curated and edited using the BioEdit v7.2.5 (Hall, 1999). Subsequently, a ML (maximum likelihood) phylogenetic tree of viral genomes was constructed using the IQ-TREE multicore version 1.6.12 (http://www.iqtree.org/release/v1.6.12) based on 1000 bootstrap replicates and best-fitting model TIM2 + F + I + G4 (Trifinopoulos et al., 2016), while the tree was visualized and modified using the FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/). The final dataset of the Maximum Likelihood method consisted of 6981 positions. The ML phylogenetic tree of viral amino acid sequences was constructed using the IQ-TREE multicore version 1.6.12 based on 1000 bootstrap replicates and best-fitting model JTTDCMut + I + G4. In addition, the genetic similarity analysis was performed using the SimPlot v3.5.
Phylogeographic Network Analysis
In order to map the regional level distribution and genetic relationships of the full-length genome sequences, a minimum spanning network (MSN) was applied using the PopArt v4.8 (https://popart.maths.otago.ac.nz/) (Leigh & Bryant, 2015). The MSN phylogeographic network included all the full-length genome sequences used in the phylogenetic tree construction, isolated from twenty-six different countries of the world (Table 1).
Amino Acids Variability Analysis
The full-length polyprotein sequences of all the full-length genomes of Echoviruses were separately retrieved from the NCBI GenBank database and were manually translated into amino acid sequences using the MEGA11 software. The amino acids variability across the full-length polyprotein was determined using the Wu-Kabat variability coefficient implemented by the Protein Variability Server (PVS, http://imed.med.ucm.es/PVS/) (Garcia-Boronat et al., 2008). The Wu-Kabat variability coefficient was calculated using the formula V = N*k/n, where V is the variability, n is the frequency that the most commonly recognized amino acid at that position is available, k represents the number of different amino acids at a given position and N is the number of sequences in the alignment (Kabat et al., 1977).
Recombination Analysis
The 277 full-length nucleotide sequences of the Echovirus were examined using Recombination Detection Program 4 software (RDP4, http://web.cbio.uct.ac.za/~darren/rdp.html) for recombination events (Martin et al., 2015). The recombination events were identified using each of the seven algorithms, including GENECONV, RDP, SiScan, Bootscan, PhylPro, MaxChi, and Chimaera embedded in the RDP4 package. A real recombination event was accepted when supported by at least four of the aforementioned algorithms.
Results
Phylogenetic Tree of Echoviruses
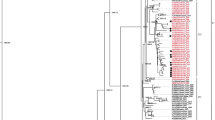
Our ML phylogenetic tree of viral genomes constructed using the best-fitting model TIM2 + F + I + G4 in IQ-TREE v1.6.12 (Trifinopoulos et al., 2016) indicated that all the 277 full-length genome sequences are classified into nine major clades, e.g., G1–G9 (Fig. 1). The phylogenetic tree indicated high diversity, where the G1 was the largest clade, clustering all the E11 and three of E19 strains (a total of 69), where most of the strains were isolated in China and Israel (25 each) (Fig. 1, Table 1, Fig. S1). Similarly, the G2 clade clustered the E7, G3 clustered E3 and E12, G4 clustered E30, E21, and E25, G5 clustered the E6 and E29, G7 clustered E9, E14 and E16, while G8 clade was limited to E18 strains only. On the other hand, the G6 and G9 clades were highly diverse, where the G6 clustered the E1, E4, E13, E20, E24, and E33, while the G9 clustered the E2, E5, E15, E17, E31, and E32. Based on the geographic locations of isolates, the strains from China and USA indicated the highest diversity and were spread across almost all the clades (Fig. 1, Table 1, Fig. S1).

Phylogenetic analysis of Echovirus based on full-length genomes, 1992–2019. A Maximum Likelihood (ML) phylogenetic tree of 277 full-length genome sequences was inferred using the IQ-TREE v1.6.12 with best-fitting model TIM2 + F + I + G4 and 1000 bootstraps. All the strains are clustered into nine major clades G1–G9. The strains isolated in China are indicated with red, USA with green and Israel with yellow color. The tree was visualized and modified using FigTree v1.4
The 22 E11 strains from China in the G1 clustered with E11-2017-122-R2 (GenBank ID: MK791152.1, China-2018) with a bootstrap value of 100%. E11-GWCMC01_GZ_CHN_2019 (GenBank ID: MN817130.1, China-2019) and E11-SD2003-478_SD_CHN_2003 (GenBank ID: MN496161.1, China-2013) strains were distant from the rest of the E11 strains from China (Fig. S1), and were clustered with the above E11 strains, with a 99% bootstrap value. The E3-Env-2016-Sep-E-3 (GenBank ID: MG451804.1, United_Kingdom-2016) strain in the G3 branch clustered with all Japanese sequences with a bootstrap value of 100% (Fig. S1). In addition, the E3 strain from China clustered with the E12 strain with a 100% bootstrap value. Importantly, two of the strains, e.g., E26 and E27 that were isolated in USA during 2003 (GenBank IDs: AY302550.1 and AY302551.1 respectively) appeared to be forming a new clade (bootstrap values 100%), but not classified. In the G7 clade, the E9 strain MSH_KM812_2010 (GenBank ID: JN596587.1) identified in China in 2010 was associated with viruses previously identified in Finland and Australia from 2002 to 2012. In addition, the USA strains identified in 2016 (GenBank ID: KX610685.2 and KX681481.1) clustered together with the USA strain (GenBank ID: MH752986.1) identified in 2004 with a bootstrap value of 89%. The E16 strains in this clade clustered together with the E14 strains with a bootstrap value of 87%. All E18 strains clustered in the G8, where USA_2015_CA-RGDS-1049 (GenBank ID: MN166092.1, USA-2015) strain clustered with all China strains with a bootstrap value of 100% (Fig. S1).
Furthermore, 277 full-length polyprotein sequences were obtained and were analyzed to construct amino acid sequence-based ML phylogenetic tree using the best-fitting model JTTDCMut + I + G4 in IQ-TREE v1.6.12 (Trifinopoulos et al., 2016). As shown in Fig. 2, the results are in consistence with that of genomic sequence-based ML phylogenetic tree (Fig. 1) with the exception of a few of virus strains. The G1 clade still clustered all E11 and three E19 strains (Fig. 2 and Fig. S2). Similarly, the G2 clade clustered E7 strains; G3 clustered E3 and E12; G4 clustered E30, E21, and E25; G5 clustered the E6 and E29; G6 clustered the E1, E4, E13, E20, E24, and E33; G8 clade was limited to E18 strains only.

Phylogenetic analysis of Echovirus based on the amino acid sequence of full-length polyprotein, 1992–2019. A Maximum Likelihood (ML) phylogenetic tree of 277 full-length polyproteins based on the amino acid sequence was inferred using the IQ-TREE v1.6.12 with best-fitting model JTTDCMut + I + G4 and 1000 bootstraps. The strains isolated in China are indicated with red, USA with green and Israel with yellow color. The tree was visualized and modified with FigTree v1.4. The genotypes were defined based on the full-length genomic sequences analyzed in Fig. 1
On the other hand, while the G9 clustered the E2, E5, E15, E17, E31, and E32 in genomic sequence-based ML phylogenetic tree (Fig. 1), the polyprotein sequence of E32 was closer to that of G2 (Fig. 2); polyproteins of E2 and E15 were closer to that of G9; polyproteins of E5 and E31 were closer to that of G7 strains (Fig. 2). In addition, both E26 and E27 are represented by only one virus strain each and form a separate clade as unclassified strains in genomic sequence-based ML phylogenetic tree (Fig. 1). However, the polyprotein of E26 was closer to that of G7 strains, but the polyprotein of E27 was sorted into an independent clade with that of E17 (Fig. 2). The inconsistent phenomenon may indicate the existence of genomic recombination.
In consistence with the phylogenetic analysis, the genetic similarity map also showed high diversity, especially at the genome position 1–3200, encoding the VP4, VP2, VP3 and VP1 proteins (similarity < 90%). The most variable region was indicated to be the nucleotide position 2500–3000 that codes for the VP1 protein, while the nucleotide position 3750–7427 that codes for the 2B, RNA helicase, 3A, C3 peptidase and the RNA dependent RNA polymerase (RdRp) were indicated to be the most conserved region of the Echovirus genome (Fig. 3).

Genetic similarity map of the full-length genome sequences of representative Echovirus strains. A Schematic diagram of Echovirus complete genome structure. From 5′ end to 3′ end are the overlapping ORFs that encodes for the VP4, VP2, VP3, VP1, P2A, P2B, RNA helicase, P3A, C3 peptidase and RdRp proteins. B SimPlot similarity analysis results using the E2_Cornelis (GenBank ID: AF465518.1, Sweden-2002) strain within the G9 as the query sequence to compare with the thirteen representative strains from other 8 clades
Phylogeographic Network of Echoviruses
The phylogeographic network was analyzed using the MSN (minimum spanning network) method (Leigh & Bryant, 2015) to further evaluate the genetic relationships and regional level spread of Echovirus in the context of full-length genomes. In agreement with our phylogenetic and genetic similarity analyses, the phylogeographic network also indicated great diversity, consisting of several mutation branches (Fig. 4). Interestingly, the clades G2–G9 are connected to three strains, e.g., E30-14-397 (GenBank ID: KY888274.1, Germany-2013), E24-DeCamp (GenBank ID: AY302548.1, USA-2003) and E30_Zhejiang_17_03_CSF (GenBank ID:DQ246620.1, China-2005), while the biggest clade G1 is connected to the G5 clade through long and inter-connecting strains between the E6SD11CHN (GenBank ID: JX976771.1, China-2011) and the E11-04_GZ_CHN_2019 (GenBank ID: MW883613.1, China-2019) strains.

Phylogeographic network analysis based on full-length genome sequences of Echovirus, 1992–2019. The phylogeographic analysis of 277 full-length genome sequences of Echovirus was performed using the Minimum Spanning Network (MSN) implemented by PopArt v1.7. The analysis showed that the G2–G7 clades are connected, while the G1 clade is connected to G5. The E12, E14 and E16 strains clustered separately from their clades G3 and G7 as a separate branch. Each node represents one strain while the distance among the nodes represents the mutational steps. Each color represents a different country
Importantly, the G3 and G7 clades are connected to a single strain E3-HNWY-01 (GenBank ID: KM388538.1, China-2012). However, four of the E12 strains within the G3 clade, e.g., E12-K605_YN_CHN_2013 (GenBank ID: MF083152.1, China-2013), E12-K1529_YN_CHN_2013 (GenBank ID: MF083154.1, China-2013), E12-K624_YN_CHN_2013 (GenBank ID: MF083153.1, China-2013) and E12-prototype-Travis (GenBank ID: X77708.1 Germany-1997), and the E14 and E16 serotypes within the G7 clade clustered as a separate mutation branch within the network. In addition, the unclassified strains E26 and E27 along with the E13-Del-Carmen strain (GenBank ID: AY302539.1, USA-2003) and the E13-GHA_VOL_KRN_2017 (GenBank ID: MH005792.1, Ghana-2017) of the G6 clade appeared as a separate branch, which were connected to the E11-GHA_UER_PUS_2017 (GenBank ID: MH005790.1, Ghana-2017) of the G1 clade (Fig. 4). These results speculate potential genetic exchange among the Echovirus strains, which lead to the rise of new clades and sub-clades.
Amino Acid Variability Landscape
As the Echovirus genomes indicated high diversity and possibility of potential genetic exchange, we further evaluated their possible effects on the amino acids’ conservancy of the Echovirus. We used the Wu-Kabat Variability coefficient method implemented by the PVS (Garcia-Boronat et al., 2008), to determine the amino acid variability landscape of the polyprotein of the Echovirus. The polyprotein was 2221 amino acids in length (Fig. 5A, B, C). The variability analysis indicated that the highest variability was shown by the aa positions 201–249, 653–827 and 400–443 that encode the VP2, VP1 and VP3 respectively (Fig. 5A), followed by the aa position 842–900 that encode the upstream and N-terminus of P2A protein (Fig. 5B). On the other hand, the aa position 1500–2221 that encodes the C3 peptidase and RNA dependent RNA polymerase (RdRp) were the most conserved region (Fig. 5C). The highest variable region identified was at aa 842–900 (highest recorded value = 65) in the P2A region (Fig. 5B). These results indicate that the structural capsid proteins of Echovirus have varied greatly during 1992–2019.

The amino acid variability landscape of Echovirus polyprotein. The amino acid variability of Echovirus polyprotein containing (A) VP4, VP2, VP3 and VP1, (B) VP1, P2A, P2B, RNA helicase and P3A, and (C) C3 peptidase and RdRp proteins was determined using the Wu-Kabat variability coefficient implemented by the PVS. The X axes represents the amino acid position, while the Y axes represents the variability coefficient, where the estimation limit is 1. The values within the estimation limit represents the conserved, while above the limit represents the variability
Recombination Pattern of Echovirus Full-Length Genome
As phylogeographic network analyses indicated possible genetic exchange, we used the full-length sequences to assess the occurrence of recombination between Echovirus strains. We performed recombination analysis of 277 full-length Echovirus genomes using seven algorithms embedded in RDP4 and identified a total of 93 recombination events (Table S1), where six were intra-clades (Events 9, 11, 37, 52, 65 and 85), while remaining were inter-clade recombination. In particular, 25 recombination events occurred within the G4, 21 within G5, 11 within G8, 8 within G1, G3 and G7 respectively, 6 within G6, 4 within G9 and 2 within G2. The E30 (G4) and E6 (G5) were the most active serotypes that appeared as recombinants in 17 events each, e.g., E30 in Events 3, 8, 11, 13, 14, 20, 33, 36, 37, 43, 56, 74, 77, 80, 81, 89 and 92, while E6 in Events 2, 12, 16, 23, 28, 31, 35, 40, 41, 57, 60, 62, 70, 71, 73, 84 and 93. Interestingly, the two strains, e.g., E26 (GenBank ID: AY302550.1) and E27 (GenBank ID: AY302551.1) that were unclassified strains in our phylogenetic analysis appeared as minor and major parents respectively for recombinants in Event 63 and 32, respectively (Table S1).
We mapped the breakpoints of the 93 Echovirus recombination events identified (Fig. S3). The majority of the recombination (a total of 27) occurred in the VP1-VP4 coding region, e.g., Events 1, 4–6, 12, 13, 15–18, 20, 29 30, 34, 38, 39, 43, 45, 47, 50, 54, 59–61, 74, 86 and 89. Similarly, 20 recombination events encompassed the RNA helicase, P3A, C3 peptidase and RdRp regions, e.g., Events 2, 3, 7, 8, 11, 14, 19, 21, 25, 26, 33, 42, 46, 48, 51, 57, 62, 68, 83 and 85, while six events encompassed the VP-P2A coding regions, e.g., Events 9, 10, 28, 56, 75 and 77. In addition, multiple recombination events occurred within each individual coding region. These extensive recombination events within the full-length genomes of Echoviruses further validate the rise of new variants that leads to the emergence of new clades and sub-clades.
Discussion
Echoviruses as members of the genus Enterovirus in the Picornaviridae family (Brouwer et al., 2021), make a large group within the Enteroviruses (Brouwer et al., 2021; Rao, 2015). Though, the Echoviruses are grouped into Enterovirus B (EV-B) by the ICTV, there is no classification on the clade and sub-clade levels (ICTV, 2023). The viruses are grouped by the studies reporting it. Therefore, we evaluated the phylogenetic classification, phylogeographic distribution, and the genetic and evolutionary diversity of Echovirus available in the NCBI database isolated from 1992 to 2019, using the 277 Echovirus full-length genome sequences. In our study. The phylogenetic tree and genetic similarity mapping analysis showed a high level of diversity. The results showed that based on whole genome sequences, these virulent strains were divided into nine major clades (G1–G9), consisting of G1 (E11 and E19), G2 (E7), G3 (E3 and E12), G4 (E30, E25 and E21), G5 (E6 and E29), G6 (E1, E4, E13, E20, E24, and E33), Unclassified strains (E26 and E27), G7 (E9, E14 and E16), G8 (E18), G9 (E2, E5, E15, E17, E31 and E32), with G1 being the largest clade. Table 1 shows that the largest number of strains were isolated from Asia, followed by Europe.
Currently, the genotype classification of Echoviruses is complicated, and the unified classification system is being expected. Previously, the Echoviruses were classified into different genogroups based on the VP1 nucleotide sequences. For instance, using 47 full-length sequences and 277 VP1 coding sequences retrieved from GenBank to construct a phylogenetic tree in 2010–2018, E30 was classified into eight genotypes G1–G8 (Benschop et al., 2021). Based on VP1 coding sequences of 53 strains of E12 isolated worldwide (including 8 isolated from Yunnan in 2013), E12 was classified into seven genotypes (A–G) (Liu et al., 2018). Similarly, E13 strains obtained from Pakistan were segregated with other E13 strains into two genogroups A and B. The strains of genogroups A and B were further clustered into A1–A7 and B1–B3 (Shaukat et al., 2017). Based on VP1 coding sequence, 73 E14 strains isolated from Shandong with other global E14 sequences, E14 was divided into lineage I and II, where all sequences in lineage I and lineage II were further divided into A–C and D–F (Chen et al., 2017a). The E16 strains collected from March to June 2019 at Mayotte Hospital and from GenBank database were divided into two branches (I and II) (Fourgeaud et al., 2022). All collected E18 strains were divided into three genotypes A, B and C. Sub-genotypes of genotype C could be further separated into C1 and C2 (Chen et al., 2017b). E19 strains were isolated from Pakistan from January to December 2013, all the strains studied constituted a separate branch and were further divided into two groups (I and II) (Angez et al., 2015). In this study, we searched the NCBI GenBank database for 277 available full-length genome sequences of Echoviruses isolated between 1992 and 2019 and have grouped all the available Echovirus serotypes into nine genogroups (G1–G9) based on the full-length genome sequences, which provides an updated and robust classification of all the Echovirus serotypes.
Previously, the phylodynamic analyses were used to determine the epidemic episodes in transmission and evolution of viruses (Grenfell et al., 2004). In our study, the phylogeographic network analysis showed a large diversity of Echovirus, where the E11 made the largest cluster (G1) within the network. E11 is reported to be associated with several outbreaks in neonatal nurseries (Abzug, 2004; Chen et al., 2005), and is the second most common according to the enterovirus surveillance conducted during 1970–2005 in USA (Khetsuriani et al., 2006). Our analysis revealed that the G2-G9 clades were linked to three common strains, where two of the strains were of E30, e.g., E30_Zhejiang_17_03_CSF-China-2005 (GenBank ID: DQ246620.1) and E30-14-397-Germany-2013 (GenBank ID: KY888274.1). Previously, it is reported that the E30 MRCA (most recent common ancestor) began to diversify in 1934, which was associated with the wide geographic dispersion possibly resulted from frequent human movement since that time (Lema et al., 2019). These results are also in consistent with our recombination analysis, where the E30 and E6 appeared in the highest number of recombination events. In addition, the biggest clade (G1) clustering the E11 strains was determined to be linked with the G5 clade (clustering the E6), which further supports the central role of E30 and E6 in the evolution of echovirus serotypes. Furthermore, we found that E26 and E27 strains (GenBank IDs: AY302550.1 and Y302551.1) appeared as separate clade (Unclassified in this report) and were connected to the E13-Del-Carmen strain (GenBank ID: AY302539.1). These results are in consistence with the previous report, suggesting that E26, E27 and E13 strains exhibit complex relationships with one another, where E26 and E27 were highly similar in 3C and 3D coding region, E13 is closer to E26 in 2C coding region and to E27 in 3AB coding region, while both E26 and E27 are closer to E13 in the 5′ end of 3D coding regions (Oberste et al., 2004).
The recombination and mutation have been suggested as the main mechanisms of Echovirus evolution (Lukashev et al., 2005). Echovirus recombination results from the exchange of a large part of the genome among different RNA strands, with study finding that P2 and P3 coding sequences are the main regions of recombination (Kyriakopoulou et al., 2015b). However, we found that the VP1–VP4 coding region has the highest recombination frequency during 1992–2019. In addition to the double recombinant virus mentioned here, a mosaic triple-recombinant between E30, E6, and E11 was reported in New Zealand in 2017 (Dilcher et al., 2022). Our results further revealed the extensive recombination between different serotypes with far genetic distance (Table S1). On the other hand, the high frequency of nucleotide misincorporation results in an increase in genetic drift of Echovirus (Rueca et al., 2022). We found that the structural capsid proteins VP2, VP1 and VP3 are highly susceptible to the amino acid variation, which contributed greatly with the recombination to the emerging of new lineages of Echoviruses, and also may affect the host determination and clinical phenotypic outcome. In picornaviruses, VP proteins play an essential role during virus replication (Fields et al., 2007). The VP1-3 proteins are located at the surface of the viral capsid and are exposed to immune pressure, while the VP4 is located inside the viral capsid (Racaniello, 2007). In many enteroviruses like EV71, the VP1-3 proteins are essential for the ability of the virus to infect the host cells and serve as protective protein in viral shell (Zhang et al., 2010). In addition, these proteins are known as major antigens in the host (Zhang et al., 2010). Thus, these variations may lead to further changes in the antigenicity of the virus, which need to be further investigated to better understand the virus life cycle and pathogenicity and assist in formulating prevention and control measures.
In summary, based on the full-length genomic sequence of Echovirus, we outline an updated phylogenetic and phylogeographic landscape to further elucidate the global distribution and classification of prevalent strains. Echovirus is globally widespread, with distribution on all continents. The rapid evolution of Echovirus is being driven by the combined forces of capsid protein mutation and extensive genomic recombination.
Data Availability
The nucleotide and protein sequence data used in this study are available on the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/nuccore/).
References
Abzug, M. J. (2004). Presentation, diagnosis, and management of enterovirus infections in neonates. Pediatric Drugs, 6, 1–10.
Angez, M., Shaukat, S., Zahra, R., Sharif, S., Alam, M. M., Khurshid, A., Rana, M. S., & Zaidi, S. S. (2015). Identification of new genotype of Echovirus 19 from children with Acute Flaccid Paralysis in Pakistan. Scientific Reports, 5, 17456.
Benschop, K. S. M., Broberg, E. K., Hodcroft, E., Schmitz, D., Albert, J., Baicus, A., Bailly, J. L., Baldvinsdottir, G., Berginc, N., Blomqvist, S., Böttcher, S., Brytting, M., Bujaki, E., Cabrerizo, M., Celma, C., Cinek, O., Claas, E. C. J., Cremer, J., Dean, J., … Demchyshyna, I. (2021). Molecular epidemiology and evolutionary trajectory of emerging Echovirus 30, Europe. Emerging Infectious Diseases, 27, 1616–1626.
Brouwer, L., Moreni, G., Wolthers, K. C., & Pajkrt, D. (2021). World-wide prevalence and genotype distribution of Enteroviruses. Viruses, 13, 434.
Castro, C. M., Oliveira, D. S., Macedo, O., Lima, M. J., Santana, M. B., Wanzeller, A. L., Silveira, E., & Gomes, M. L. (2009). Echovirus 30 associated with cases of aseptic meningitis in state of Pará, Northern Brazil. Memorias Do Instituto Oswaldo Cruz, 104, 444–450.
Chen, J. H., Chiu, N. C., Chang, J. H., Huang, F. Y., Wu, K. B., & Lin, T. L. (2005). A neonatal echovirus 11 outbreak in an obstetric clinic. Journal of Microbiology, Immunology, and Infection, 38, 332–337.
Chen, P., Li, Y., Tao, Z., Wang, H., Lin, X., Liu, Y., Wang, S., Zhou, N., Wang, P., & Xu, A. (2017a). Evolutionary phylogeography and transmission pattern of echovirus 14: An exploration of spatiotemporal dynamics based on the 26-year acute flaccid paralysis surveillance in Shandong, China. BMC Genomics, 18, 48.
Chen, X., Li, J., Guo, J., Xu, W., Sun, S., & Xie, Z. (2017b). An outbreak of echovirus 18 encephalitis/meningitis in children in Hebei Province, China, 2015. Emerging Microbes & Infections, 6, e54.
Chen, X., Qu, X., Liu, C., Zhang, Y., Zhang, G., Han, P., Duan, Y., Li, Q., Wang, L., Ruan, W., Wang, P., Wei, W., Gao, G. F., Zhao, X., & Xie, Z. (2022). Human FcRn is a two-in-one attachment-uncoating receptor for Echovirus 18. mBio, 13, e0116622.
Diedrich, S., & Schreier, E. (2001). Aseptic meningitis in Germany associated with echovirus type 13. BMC Infectious Diseases, 1, 14.
Dilcher, M., Howard, J. C., Dalton, S. C., Anderson, T., Clinghan, R. T., & Werno, A. M. (2022). Clinical, laboratory, and molecular epidemiology of an outbreak of aseptic meningitis due to a triple-recombinant Echovirus in Ashburton, New Zealand. Viruses, 14, 658.
Faleye, T. O. C., Adewumi, O. M., Klapsa, D., Majumdar, M., Martin, J., & Adeniji, J. A. (2019). Genome Sequences of two dual-serotype-specific Echovirus 20 Strains from Nigeria. Microbiology Resource Announcements, 8, e00894-e919.
Fields, B. N., Knipe, D. M., & Howley, P. M. (2007). Fields virology (5th ed.). Wolters Kluwer Health/Lippincott Williams & Wilkins.
Fourgeaud, J., Mirand, A., Demortier, J., Kamus, L., Collet, L., Olivier, S., Henquell, C., & Vauloup-Fellous, C. (2022). Enterovirus meningitis in Mayotte French Comoros Island, March–June 2019. Journal of Clinical Virology, 150–151, 105154.
Fratty, I. S., Kriger, O., Weiss, L., Vasserman, R., Erster, O., Mendelson, E., Sofer, D., & Weil, M. (2023). Increased detection of Echovirus 6-associated meningitis in patients hospitalized during the COVID-19 pandemic, Israel 2021–2022. Journal of Clinical Virology, 162, 105425.
Garcia-Boronat, M., Diez-Rivero, C. M., Reinherz, E. L., & Reche, P. A. (2008). PVS: A web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Research, 36, W35–W41.
Grädel, C., Ireddy, N. R., Koch, M. C., Baumann, C., Terrazos Miani, M. A., Barbani, M. T., Steinlin-Schopfer, J., Suter-Riniker, F., Leib, S. L., & Ramette, A. (2022). Genome sequences of rare human enterovirus genotypes recovered from clinical respiratory samples in Bern. Switzerland. Microbiology Resource Announcements, 11, e0027622.
Grenfell, B. T., Pybus, O. G., Gog, J. R., Wood, J. L., Daly, J. M., Mumford, J. A., & Holmes, E. C. (2004). Unifying the epidemiological and evolutionary dynamics of pathogens. Science, 303, 327–332.
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Handsher, R., Shulman, L. M., Abramovitz, B., Silberstein, I., Neuman, M., Tepperberg-Oikawa, M., Fisher, T., & Mendelson, E. (1999). A new variant of echovirus 4 associated with a large outbreak of aseptic meningitis. Journal of Clinical Virology, 13, 29–36.
Hirade, T., Abe, Y., Ito, S., Suzuki, T., Katano, H., Takahashi, N., Koike, D., Nariai, A., & Kato, F. (2023). Congenital Echovirus 11 infection in a neonate. The Pediatric Infectious Disease Journal. https://doi.org/10.1097/inf.0000000000004052
Home of Picornaviruses. (2023). Enterovirus B. The Pirbright Institute. Retrieved from https://www.picornaviridae.com/ensavirinae/enterovirus/ev-b/ev-b.htm.
Huang, Q. S., Carr, J. M., Nix, W. A., Oberste, M. S., Kilpatrick, D. R., Pallansch, M. A., Croxson, M. C., Lindeman, J. A., Baker, M. G., & Grimwood, K. (2003). An echovirus type 33 winter outbreak in New Zealand. Clinical Infectious Diseases, 37, 650–657.
ICTV, International Committee on Taxonomy of Viruses. (2023). Taxon Details. Enterovirus B. Retrieved from https://ictv.global/taxonomy/taxondetails?taxnode_id=202101984 (20 Feb 2023).
Jiang, C., Xu, Z., Li, J., Zhang, J., Xue, X., Jiang, J., Jiang, G., Wang, X., Peng, Y., Chen, T., et al. (2022). Case report: Clinical and virological characteristics of aseptic meningitis caused by a recombinant echovirus 18 in an immunocompetent adult. Frontiers in Medicine, 9, 1094347.
Kabat, E. A., Wu, T. T., & Bilofsky, H. (1977). Unusual distributions of amino acids in complementarity determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. Journal of Biological Chemistry, 252, 6609–6616.
Khetsuriani, N., Lamonte-Fowlkes, A., Oberst, S., Pallansch, M. A., Centers for Disease Control and Prevention. (2006). Enterovirus surveillance—United States, 1970–2005. Morbidity and Mortality Weekly Report. Surveillance Summaries, 55, 1–20.
Kyriakopoulou, Z., Bletsa, M., Tsakogiannis, D., Dimitriou, T. G., Amoutzias, G. D., Gartzonika, C., Levidiotou-Stefanou, S., & Markoulatos, P. (2015a). Molecular epidemiology and evolutionary dynamics of Echovirus 3 serotype. Infection, Genetics and Evolution, 32, 305–312.
Kyriakopoulou, Z., Pliaka, V., Amoutzias, G. D., & Markoulatos, P. (2015b). Recombination among human non-polio enteroviruses: Implications for epidemiology and evolution. Virus Genes, 50, 177–188.
Lavania, M., Viswanathan, R., Bhardwaj, S. D., Oswal, J. S., Chavan, N., Shinde, M., & Katendra, S. (2022). Detection of Echovirus-18 in children suspected With SARS-CoV-2 infection with multisystem inflammatory syndrome: A case report from India. Frontiers in Public Health, 10, 897662.
Leigh, J. W., & Bryant, D. (2015). POPART: Full-feature software for haplotype network construction. Methods in Ecology and Evolution, 6, 1110–1116.
Lema, C., Torres, C., Van der Sanden, S., Cisterna, D., Freire, M. C., & Gómez, R. M. (2019). Global phylodynamics of Echovirus 30 revealed differential behavior among viral lineages. Virology, 531, 79–92.
Li, J., Yan, D., Chen, L., Zhang, Y., Song, Y., Zhu, S., Ji, T., Zhou, W., Gan, F., Wang, X., Hong, M., Guan, L., Shi, Y., Wu, G., & Xu, W. (2019). Multiple genotypes of Echovirus 11 circulated in mainland China between 1994 and 2017. Scientific Reports, 9, 10583.
Liu, H., Zhang, J., Zhao, Y., Zhang, H., Lin, K., Sun, H., Huang, X., Yang, Z., & Ma, S. (2018). Molecular characterization of echovirus 12 strains isolated from healthy children in China. Scientific Reports, 8, 11716.
Lizasoain, A., Mir, D., Victoria, M., Barrios, M. E., Blanco-Fernández, M. D., Rodríguez-Osorio, N., Nates, S., Cisterna, D., Mbayed, V. A., & Colina, R. (2021). Human Enterovirus diversity by next-generation sequencing analysis in urban sewage samples from Buenos Aires Metropolitan Area, Argentina: A retrospective study. Food and Environmental Virology, 13, 259–269.
Lukashev, A. N., Lashkevich, V. A., Ivanova, O. E., Koroleva, G. A., Hinkkanen, A. E., & Ilonen, J. (2005). Recombination in circulating Human enterovirus B: Independent evolution of structural and non-structural genome regions. The Journal of General Virology, 86, 3281–3290.
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., & Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evolution, 1, vev003.
Miyata, I., Hanaoka, N., Okabe, N., Fujimoto, T., Sakamoto, S., Kasahara, M., & Saitoh, A. (2014). Echovirus 3 as another enterovirus causing life-threatening neonatal fulminant hepatitis. Journal of Clinical Virology, 59, 132–134.
Morsli, M., Vincent, J. J., Milliere, L., Colson, P., & Drancourt, M. (2021). Direct next-generation sequencing diagnosis of echovirus 9 meningitis, France. European Journal of Clinical Microbiology & Infectious Diseases, 40, 2037–2039.
Oberste, M. S., Maher, K., & Pallansch, M. A. (2004). Evidence for frequent recombination within species Human Enterovirus B based on complete genomic sequences of all thirty-seven serotypes. Journal of Virology, 78, 855–867.
Oberste, M. S., Nix, W. A., Kilpatrick, D. R., Flemister, M. R., & Pallansch, M. A. (2003). Molecular epidemiology and type-specific detection of echovirus 11 isolates from the Americas, Europe, Africa, Australia, southern Asia and the Middle East. Virus Research, 91, 241–248.
Ogbole, O. O., Akinleye, T. E., Segun, P. A., Faleye, T. C., & Adeniji, A. J. (2018). In vitro antiviral activity of twenty-seven medicinal plant extracts from Southwest Nigeria against three serotypes of echoviruses. Virology Journal, 15, 110.
Paul, A. V., & Wimmer, E. (2015). Initiation of protein-primed picornavirus RNA synthesis. Virus Research, 206, 12–26.
Pedrosa, C., Lage, M. J., & Virella, D. (2013). Congenital echovirus 21 infection causing fulminant hepatitis in a neonate. BMJ Case Reports. https://doi.org/10.1136/bcr-2012-008394
Racaniello, V. R. (2007). Picornaviridae: The viruses and their replication. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, & S. E. Strauss (Eds.), Fields virology (5th ed., pp. 795–838). Lippincott Williams & Wilkins.
Ramos-Alvarez, M., & Sabin, A. B. (1954). Characteristics of poliomyelitis and other enteric viruses recovered in tissue culture from healthy American children. Proceedings of the Society for Experimental Biology and Medicine, 87, 655–661.
Rao, C. D. (2015). Non-polio enteroviruses, the neglected and emerging human pathogens: Are we waiting for the sizzling enterovirus volcano to erupt. Proceedings of the Indian National Science Academy, 81, 447–462.
Roberts, J. A., Hobday, L. K., Ibrahim, A., & Thorley, B. R. (2020). Australian National Enterovirus Reference laboratory annual report 2018. Communicable Diseases Intelligence. https://doi.org/10.33321/cdi.2020.44.26
Rueca, M., Lanini, S., Giombini, E., Messina, F., Castilletti, C., Ippolito, G., Capobianchi, M. R., & Valli, M. B. (2022). Detection of recombinant breakpoint in the genome of human enterovirus E11 strain associated with a fatal nosocomial outbreak. Virol Journal, 19, 97.
Shaukat, S., Angez, M., Mahmood, T., Alam, M. M., Sharif, S., Khurshid, A., Rana, M. S., & Zaidi, S. S. (2017). Molecular characterization of echovirus 13 uncovering high genetic diversity and identification of new genotypes in Pakistan. Infection, Genetics and Evolution, 48, 102–108.
Sousa, I. P., Jr., Burlandy, F. M., Lima, S. T. S., Maximo, A. C. B., Figueiredo, M. A. A., Maia, Z., & da Silva, E. E. (2019). Echovirus 30 detection in an outbreak of acute myalgia and rhabdomyolysis, Brazil 2016–2017. Clinical Microbiology and Infection, 25, 252.e2-252.e8.
Sun, Y., Miao, Z., Yan, J., Gong, L., Chen, Y., Chen, Y., Mao, H., & Zhang, Y. (2019). Sero-molecular epidemiology of enterovirus-associated encephalitis in Zhejiang Province, China, from 2014 to 2017. International Journal of Infectious Diseases, 79, 58–64.
Tamura, K., Stecher, G., & Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Molecular Biology and Evolution, 38, 3022–3027.
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., & Minh, B. Q. (2016). W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Research, 44, W232–W235.
Wieczorek, M., Krzysztoszek, A., Ciąćka, A., & Figas, A. (2017). Molecular characterization of environmental and clinical echovirus 6 isolates from Poland, 2006–2014. Journal of Medical Virology, 89, 936–940.
Xi, H., Tian, Y., Shao, H., Yin, X., Ma, L., Yang, P., & Li, X. (2023). An outbreak of nosocomial infection of neonatal aseptic meningitis caused by echovirus 18. Epidemiology and Infection, 151, e107.
Xiao, J., Zhang, Y., Hong, M., Han, Z., Zhang, M., Song, Y., Yan, D., Zhu, S., & Xu, W. (2020). Phylogenetic characteristics and molecular epidemiological analysis of novel enterovirus EV-B83 isolated from Tibet, China. Scientific Reports, 10, 6630.
Yao, X., Bian, L. L., Mao, Q. Y., Zhu, F. C., Ye, Q., & Liang, Z. L. (2015). Echovirus 7 associated with hand, foot, and mouth disease in mainland China has undergone a recombination event. Archives of Virology, 160, 1291–1295.
Zhang, D., Lu, J., & Lu, J. (2010). Enterovirus 71 vaccine: Close but still far. International Journal of Infectious Diseases, 14, e739–e743.
Zhang, M., Guo, W., Xu, D., Feng, C., Bao, G., Sun, H., Yang, Z., & Ma, S. (2022). Molecular characterization of echovirus 9 strains isolated from hand-foot-and-mouth disease in Kunming, Yunnan Province. China. Scientific Reports, 12, 2293.
Zhu, Y., Zhou, X., Liu, J., Xia, L., Pan, Y., Chen, J., Luo, N., Yin, J., & Ma, S. (2016). Molecular identification of human enteroviruses associated with aseptic meningitis in Yunnan province, Southwest China. Springerplus, 5, 1515.
Acknowledgements
This work is supported by Four “batches” innovation project of invigorating medical through science and technology of Shanxi province for Dr. Li Xing (2023XM015), the Fundamental Research Program of Shanxi Province, China (20210302124187) for Dr. Yue Liu, and the Programme of Introducing Talents of Discipline to Universities (D21004). We thank all the persons who contributed to the collection and production of Echovirus sequences in NCBI GenBank.
Author information
Authors and Affiliations
Contributions
LX conceived the study. YW, PTS, and YL performed analysis. YW, PTS, and YL wrote the manuscript. PTS, YW, ANB, and LX revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Institutional Review Board Statement and Informed Consent Statement
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, Y., Shah, P.T., Liu, Y. et al. Genetic Characteristics and Phylogeographic Dynamics of Echovirus. J Microbiol. 61, 865–877 (2023). https://doi.org/10.1007/s12275-023-00078-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12275-023-00078-w