
Abstract
Using first-principles method, for h-BN bilayer, we successfully probe the major factors of different low-friction paths in the three-dimensional potential energy surface (3D-PES) under variable loads. By means of the static PES and charge density difference analysis, we demonstrate how electrostatic interactions, with regard for van der Waals contributions at 0 nN, progressively impact the shape of 3D-PES and low-friction paths with increasing the normal load. Herein, the sliding properties of h-BN bilayers have a distinct relative orientation. Especially, the load-induced 3D-PES with variable shape is assigned to the band gap and repulsive van der Waals force. It is noted that the low friction not only is obtained for the commensurate layers under low loads, but also high ones.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Hexagonal boron nitride (h-BN), which has a structural analogue of graphene, has attracted extensive attention due to its outstanding properties [1]. These superior chemical and physical properties impel us continuously to exploit the cause that h-BN exhibits superior performance to the bulk counterpart applied in transistor, solid lubricant, high-temperature environment, and energy storage [2,3,4]. Moreover, researchers have particularly payed more attentions to monolayer h-BN (the so-called white graphene [5]) due to its high in-plane mechanical strength, thermal conductivity and chemical stability, and distinct oxidation resistance performance compared to graphene [5,6,7,8]. These exceptional properties of monolayer h-BN indicate its potential as an anti-wear material.
Watanabe et al. report that h-BN film deposited on the silicon substrate shows a short lubricating endurance life due to its lower adhesion to the silicon substrate [9]. If h-BN film well adheres to substrate, they also believe that it is a good solid lubricant. Indeed, the monolayer h-BN as a anti-wear coating for Cu surface could well reduce the exceptional low friction as reported by Li et al. [8]. Besides, h-BN as a lubricant additive also plays an important role in enhancing wear resistance and reducing friction coefficient [10,11,12,13]. The same tribological theory could be concluded from these references [8,9,10,11,12,13,14] that h-BN well adhesion to substrate or worn surfaces is a key factor to obtain high wear resistance and low-friction coefficient during sliding. Thus, understanding the active mechanism of h-BN for the tribological improvements is conducive to probing the low-friction behavior. Given that the complex interactions during sliding process are not directly monitored in experiments at the nanoscale, first-principles method offers a powerful tool to capture atomic details and gain a deeper insight into the complex interactions at the nanoscale. Accordingly, Koskilinna et al. systematically investigate the tribological properties of h-BN based on first-principles calculations [15]. They point out that the friction coefficient of h-BN is enhanced with increase of the normal load and low-friction coefficient can only be obtained at low normal load. Thereafter, a question is: what are the key factors for the different friction coefficients with increase of the normal load?
Herein, in order to well answer this question, the interfaces between two h-BN monolayers with different normal loads and the static three-dimensional potential energy surfaces (3D-PES) assigned to their motion to the interlayer forces are calculated using first-principles calculations. The calculated 3D-PES show that the low friction not only could be obtained for the commensurate layers under low normal load, but also high normal load. We discuss the unusually tribological behavior in detail to well probe the active mechanism.
2 Computational Details
All calculations are carried out using CASTEP code [16]. The DFT-D method is proved to be an effective tool due to the good agreement among the geometrical parameters, the experimental and high-accuracy theoretical data [17, 18]. Thus, the generalized gradient approximation (GGA) with PBE-D scheme reported by Grimme [19], which can well account for van der Waals interactions in all calculations. A kinetic cutoff energy of 400 eV is used for the plane-wave basis set. A 10 × 10 × 1 Monkhorst–Pack mesh is used for the k-point sampling. The electron density difference is used to investigate the interaction of two h-BN layers.
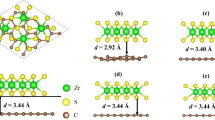
The bulk lattice parameters a and c are calculated as 2.494 and 6.488 Å, respectively, which is well in consistent with the experimental data (2.503 and 6.661 Å) [20]. The calculated model as shown in Fig. 1 is constructed with the same monolayer h-BN. The periodic boundary conditions are set in the x–y plane, and interactions between periodic replicas are avoided by adding 15 Å of vacuum along the z direction. With the upper h-BN monolayer translation above the lower one, the three-dimensional potential energy surfaces (3D-PES) is constructed by collecting the interlayer interaction energy for different relative lateral positions [21]. In particular, for Fig. 1b, along x, y, or xy direction, the h-BN bilayer coincides after the upper h-BN monolayer moving 0.5 Å above the lower one.

a Top view of a h-BN monolayer, b side view of h-BN bilayer with hollow–hollow (H–H) or top–top (T–T) configuration, c top view of h-BN bilayer with hollow–top (H–T) configuration
Next, the compressed bilayer configurations, in which the interlayer distance is fixed at different values, are considered to investigate the effect of increasing the normal load. The force along z direction as a functional of z is defined as \({F_z}= - \frac{{\partial V(z)}}{{\partial z}},\) where V(z) is the potential energy with different interlayer distances along z direction.
3 Results
As shown in Fig. 2, the potential energy and its first derivative with respect to the interlayer distance along z direction are monotonic curves. The potential energy initially kept a constant of 0 eV ranges from 4.0 to 3.5 Å, the position of the minimum energy locates at ~ 3.5 Å, which is well in consistent with previous computational results [15]. Less than 3.5 Å, the potential energy increases with decrease of interlayer distance. The variable of Fz exhibits a similar change tendency with that of potential energy.

Variable of potential energy and its first derivative with respect to the interlayer distance along z direction
Next, to well investigate the effect of the normal load on sliding properties of h-BN bilayer, the three-dimensional potential energy surfaces (3D-PES) for H–H (T–T) configuration are calculated with different normal loads as shown in Fig. 3. In Fig. 3a, under the normal load of 0 nN, there exists low-friction path along X or Y direction, however, increasing the normal load to 1.4 nN, only one low-friction path is observed along Y direction, the low-friction path along Y direction is blocked under high normal load, as shown in Fig. 3b. Interesting, further rising the normal load to 6.0 nN, the obvious low-friction path is found along X direction (Fig. 3c). It indicates that the low friction of h-BN bilayer can be achieved at high normal load for H–H (T–T) configuration.

Three-dimensional potential energy surfaces for the sliding motion of h-BN bilayers in H–H (T–T) configuration at a 0 nN, b 1.4 nN, and c 6.0 nN
For comparison, the potential energy and its first derivative with respect to the interlayer distance along z direction for H–T configuration are shown in Fig. 4. Meanwhile, the position of the minimum energy also locates at ~ 3.5 Å. The potential energy and Fz also exhibit the same change trend compared to that of H–H (T–T) configuration. Subsequently, the three-dimensional potential energy surfaces for H–T configuration are calculated as shown in Fig. 5, under the normal load of 0 and 0.6 nN, the continuous mountain-like potential energy surfaces are observed in Fig. 5a, b, the low-friction path is obviously detected along the mountain valley (Y direction). However, increasing the normal load to 3.7 nN, the shape of PES is quite different from that in Fig. 5a, b, the low-friction paths along Y direction are completely blocked; however, the low-friction path can be obtained along the edge of X direction (low potential energy region).

Variable of potential energy and its first derivative with respect to the interlayer distance along z direction

Three-dimensional potential energy surfaces for the sliding motion of h-BN bilayers in H–T configuration at a 0 nN, b 0.6 nN, and c 3.7 nN
As we know, the tribological behavior is closely related to its electronic structure. Thus, the charge density difference of h-BN bilayer in H–H (T–T), H–T configuration is calculated as shown in Fig. 6. The mixed covalent-ionic bonding characteristics, the valence charges are located around N atoms. A common conclusion can be obtained from Fig. 6a–f that the electron clouds between B atoms in h-BN bilayer overlap each other with increase of the normal load, indicating that low-friction path can be assigned to the interactions between B atoms in h-BN bilayer.

The charge density difference of h-BN bilayer in H–H or T–T configuration at a 0 nN, b 1.4 nN, and c 6.0 nN; in H–T configuration at d 0 nN, e 0.6 nN, and f 3.7 nN
Zhang et al. reported [22] the kinetic frictional force (fx) equals the predicted \(\left( {\frac{{\partial {\sigma _{\operatorname{int} }}}}{{\partial x}}} \right)\left| {\hbox{max} ,\,{\sigma _{\operatorname{int} }}} \right.\) is interfacial energy of h-BN slabs. And they also pointed out that kinetic frictional force was closely related to the change of potential energy along the sliding direction. Thus, the kinetic frictional forces with respect to sliding distance along x direction are plotted in Fig. 7. We obtain a similar result that the kinetic frictional force is determined by the change of potential energy along x direction. Besides, there is a one-to-one correspondence between frictional force and charge density difference. It has to be noted that the so-called D disorder peak is observed in well-ordered graphene, corresponding to A edge vibration modes on both external and internal edges, which can only exist on single hexagonal ring or for cluster of reduced number of cyclic rings. Differently, we calculated Raman spectra of monolayered h-BN as shown in Fig. 8. A D peak at 1339.5 cm−1 is obviously observed corresponding to the high-frequency intra-layer E2g vibration mode [7, 23] (for detailed vibration mode, see Supplementary Movie), which is well in agreement with the experimental data (1330.2–1367.4 cm−1) [24].

Variation of frictional force along x direction: a 0 nN, b 1.4 nN, and c 6.0 nN for H–H or T–T configuration; d 0 nN, e 0.6 nN, and f 3.7 nN for H–T configuration

Calculated Raman spectra of monolayered h-BN. Inset is vibration mode of h-BN at 1339.5 cm−1
4 Discussion
Generally, the anti-wear of substrate and coating system closely depends on the adhesion between friction pairs and coating. The anti-wear results from lower friction and anti-adherence between coating and the gliding counterpart [25]. Based on Neuville’s views, we find that low friction should correspond to low adhesion between coating and the gliding counterpart. On the other hand, a D disorder peak is observed in Fig. 8, it indicates that h-BN is a similar structure compared to carbon materials. And Neuville et al. believed that h-BN could be transformed into harder c-BN with the incidence of quantum electronic activation, meanwhile c-BN could be heat-degraded to h-BN during sliding [23, 26]. Thus, the quantum electronic atomic rearrangement model has also been applied in h-BN. Thus, we systematically investigate the tribological properties of h-BN bilayer under variable normal load. We obtain a quite different conclusion from the previous work [15]. Based on Tomanek’s theory [27], the kinetic friction coefficient could be calculated by the formula \(\mu \,=\,\frac{{{F_{\text{f}}}}}{{{F_{\text{N}}}}}\), combination of Figs. 7 and 8, FN equals FvdW at 0 nN, thus, the \({\mu _{{k_{\hbox{max} }}}}\) is estimated to be about 0.75 (H–H or T–T) and 0.37 (H–T), the rest FN equals Fz minus FvdW, \({\mu _{{{\text{k}}_{\hbox{max} }}}}\) is estimated to be 1.1 and 2.4 for 1.4 and 6.0 nN (H–H or T–T), and 0.15 and 1.7 for 0.6 and 3.7 nN (H–T), respectively. It is well in agreement with the change trend of friction coefficient as reported by the previous experimental researches [14, 28, 29]. In order to well understand this strength phenomenon, we should pay more attention to its electronic structure. As we known, h-BN has a similar hexagonal structure to graphene that applied in many fields. However, h-BN is a semiconductor with wide band gap, thus it exhibits good electrical insulation. An et al. reported pressure-induced insulator-semiconductor transition in h-BN bilayer [30]. We calculate the band gap for H–H (T–T) and H–T configurations under different normal loads. The band gap is 4.23 (0 nN), 3.79 (1.4 nN), and 2.26 eV (6.0 nN) for H–H (T–T) configurations, and 4.68 (0 nN), 4.57 (0.6 nN), and 4.19 eV (3.7 nN) for H–T configurations. It indicates that the band gap of H–H (T–T) configurations sensitively decreases with increase of the normal load compared to that of H–T configurations. And the band gap has a significant effect on the sliding properties of semiconductor materials as reported in our previous work [31]. We point out that the low friction can retain under higher band gap. Thus, high band gap should be one of key factors to construct the low-friction path in 3D-PES. On the other hand, it is reported that h-BN has strong bonding in xy plane but weak bonding along z direction [20]. It implies the strong covalent interlayer bonding and weak interlayer van der Waals bonding. As increasing the normal load, the van der Waals interaction is enhanced as shown in Fig. 9, the positive values of FvdW indicate a repulsion [32] in h-BN bilayer. The strong repulsive FvdW can maintain the low friction as reported in most of previous investigations [33,34,35]. In this work, the Fz is almost balanced by Fvdw at 0 and 0.6 nN (H–T), as shown in Figs. 2, 4, and 9, ensuring its low friction. Thus, the change of charge density difference with the augmenter of the normal load leading to enhancing repulsive FvdW should be another key factor for the low-friction path in 3D-PES.

The repulsive force FvdW as a function of interlayer distance along z direction for H–H (T–T) and H–T configurations
Of course, a detailed tribological investigation should take into account numerous variables such as hardness, the sliding velocity, the compressive stress, tensile stress, and so on. Given that studying the effect of these factors on low friction beyond the scope of this work, here, we simply aim at probing the possible relationship between low friction and potential energy surfaces.
5 Conclusions
By means of first-principles method, we construct the potential energy surfaces for the sliding motion of h-BN bilayers to probe their tribological properties depending on the applied the normal load or lateral orientation. Two h-BN bilayers as H–H (T–T) and H–T configuration are established. The load-induced low-friction paths are obviously observed for two models, which indicates its better lubricant properties under high load.
We also detect the delicated interplay between electrostatic, band gap, and van der Waals contributions to the h-BN bilayer. We find that high band gap of h-BN bilayer is conducive to constructing low-friction paths in 3D-PES and electron interactions of B atoms in between monolayer h-BN accelerate the increase of the repulsive Van der Waals force maintaining low-friction paths. We believe that this investigation on h-BN provides positive guidance for further experimental investigations.
References
Yi, M., Shen, Z., Zhao, X., Liang, S., Liu, L.: Boron nitride nanosheets as oxygen-atom corrosion protective coatings. Appl. Phys. Lett. 104(14), 143101 (2014). https://doi.org/10.1063/1.4870530
Xu, M., Liang, T., Shi, M., Chen, H.: Graphene-like two-dimensional materials. Chem. Rev. 113(5), 3766–3798 (2013). https://doi.org/10.1021/cr300263a
Golberg, D., Bando, Y., Huang, Y., Terao, T., Mitome, M., Tang, C., Zhi, C.: Boron nitride nanotubes and nanosheets. ACS Nano. 4(6), 2979–2993 (2010). https://doi.org/10.1021/nn1006495
Eichler, J., Lesniak, C.: Boron nitride (BN) and BN composites for high-temperature applications. J. Eur. Ceram. Soc. 28(5), 1105–1109 (2008). https://doi.org/10.1016/j.jeurceramsoc.2007.09.005
Song, L., Ci, L., Lu, H., Sorokin, P.B., Jin, C., Ni, J., Kvashnin, A.G., Kvashnin, D.G., Lou, J., Yakobson, B.I., Ajayan, P.M.: Large scale growth and characterization of atomic hexagonal boron nitride layers. Nano Lett. 10(8), 3209–3215 (2010). https://doi.org/10.1021/nl1022139
Zhi, C., Bando, Y., Tang, C., Kuwahara, H., Golberg, D.: Large-scale fabrication of boron nitride nanosheets and their utilization in polymeric composites with improved thermal and mechanical properties. Adv. Mater. 21(28), 2889–2893 (2009). https://doi.org/10.1002/adma.200900323
Shen, H., Guo, J., Wang, H., Zhao, N., Xu, J.: Bioinspired modification of h-BN for high thermal conductive composite films with aligned structure. ACS Appl. Mater. Interfaces. 7(10), 5701–5708 (2015). https://doi.org/10.1021/am507416y
Xuemei, L., Jun, Y., Jianxin, Z., Wanlin, G.: Large area hexagonal boron nitride monolayer as efficient atomically thick insulating coating against friction and oxidation. Nanotechnology 25(10), 105701 (2014)
Watanabe, S., Miyake, S., Murakawa, M.: Tribological properties of cubic, amorphous and hexagonal boron nitride films. In: Metallurgical Coatings and Thin Films, pp. 406–410. Oxford, Elsevier (1991)
Cho, D.-H., Kim, J.-S., Kwon, S.-H., Lee, C., Lee, Y.-Z.: Evaluation of hexagonal boron nitride nano-sheets as a lubricant additive in water. Wear. 302(1), 981–986 (2013). https://doi.org/10.1016/j.wear.2012.12.059
Podgornik, B., Kosec, T., Kocijan, A., Donik, Č: Tribological behaviour and lubrication performance of hexagonal boron nitride (h-BN) as a replacement for graphite in aluminium forming. Tribol. Int. 81, 267–275 (2015). https://doi.org/10.1016/j.triboint.2014.09.011
Kumari, S., Sharma, O.P., Gusain, R., Mungse, H.P., Kukrety, A., Kumar, N., Sugimura, H., Khatri, O.P.: Alkyl-chain-grafted hexagonal boron nitride nanoplatelets as oil-dispersible additives for friction and wear reduction. ACS Appl. Mater. Interfaces. 7(6), 3708–3716 (2015). https://doi.org/10.1021/am5083232
Wei, D., Meng, Q., Jia, D.: Mechanical and tribological properties of hot-pressed h-BN/Si3N4 ceramic composites. Ceram. Int. 32(5), 549–554 (2006). https://doi.org/10.1016/j.ceramint.2005.04.010
Tyagi, R., Xiong, D., Li, J.: Effect of load and sliding speed on friction and wear behavior of silver/h-BN containing Ni-base P/M composites. Wear. 270(7), 423–430 (2011). https://doi.org/10.1016/j.wear.2010.08.013
Koskilinna, J.O., Linnolahti, M., Pakkanen, T.A.: Friction coefficient for hexagonal boron nitride surfaces from ab initio calculations. Tribol. Lett. 24(1), 37–41 (2006). https://doi.org/10.1007/s11249-006-9120-x
Clark Stewart, J., Segall Matthew, D., Pickard Chris, J., Hasnip Phil, J., Probert Matt, I.J., Refson, K., Mike, P.: First principles methods using CASTEP. Zeitschrift für Kristallographie 220, 567 (2005)
Björkman, T., Gulans, A., Krasheninnikov, A.V., Nieminen, R.M.: van der Waals bonding in layered compounds from advanced density-functional first-principles calculations. Phys. Rev. Lett. 108(23), 235502 (2012)
Zhang, R., Zhao, J., Yang, Y., Shi, W., Lu, Z., Wang, J.: Understanding the friction behavior of sulfur-terminated diamond-like carbon films under high vacuum by first-principles calculations. Curr. Appl. Phys. 18(3), 317–323 (2018). https://doi.org/10.1016/j.cap.2018.01.006
Grimme, S.: Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27(15), 1787–1799 (2006). https://doi.org/10.1002/jcc.20495
Liu, L., Feng, Y.P., Shen, Z.X.: Structural and electronic properties of h-BN. Phys. Rev. B 68(10), 104102 (2003)
Zilibotti, G., Righi, M.C.: Ab initio calculation of the adhesion and ideal shear strength of planar diamond interfaces with different atomic structure and hydrogen coverage. Langmuir. 27(11), 6862–6867 (2011). https://doi.org/10.1021/la200783a
Zhang, Q., Qi, Y., Hector, L.G., Cagin, T., Goddard, W.A.: Origin of static friction and its relationship to adhesion at the atomic scale. Phys. Rev. B. 75(14), 144114 (2007). https://doi.org/10.1103/PhysRevB.75.144114
Neuville, S.: Carbon Structure Analysis with Differentiated Raman Spectroscopy: Refined Raman Spectroscopy Fundamentals For Improved Carbon Material Engineering. Riga, LAP Lambert Academic Publishing, (2014)
Sachdev, H., Haubner, R., Nöth, H., Lux, B.: Investigation of the c-BN/h-BN phase transformation at normal pressure. Diam. Relat. Mater. 6(2), 286–292 (1997). https://doi.org/10.1016/S0925-9635(96)00697-8
Neuville, S.: Antiwear material criterial. JPJ Solids Struct 3, 33–42 (2009)
Neuville, S.: Quantum electronic mechanisms of atomic rearrangements during growth of hard carbon films. Surf. Coatings Technol. 206(4), 703–726 (2011). https://doi.org/10.1016/j.surfcoat.2011.07.055
Tománek, D., Zhong, W., Thomas, H.: Calculation of an atomically modulated friction force in atomic-force microscopy. EPL 15(8), 887 (1991)
Chen, B., Bi, Q., Yang, J., Xia, Y., Hao, J.: Tribological properties of solid lubricants (graphite, h-BN) for Cu-based P/M friction composites. Tribol. Int. 41(12), 1145–1152 (2008). https://doi.org/10.1016/j.triboint.2008.02.014
Pawlak, Z., Kaldonski, T., Pai, R., Bayraktar, E., Oloyede, A.: A comparative study on the tribological behaviour of hexagonal boron nitride (h-BN) as lubricating micro-particles—an additive in porous sliding bearings for a car clutch. Wear. 267(5), 1198–1202 (2009). https://doi.org/10.1016/j.wear.2008.11.020
An, X., Sun, J., Lu, Z., Ma, F., Zhang, G.: Pressure-induced insulator-semiconductor transition in bilayer hexagonal boron nitride. Ceram. Int. 43(8), 6626–6630 (2017). https://doi.org/10.1016/j.ceramint.2017.02.037
Zhang, R., Wang, L.: Effect of compressive strain on the Hertzian contact of self-mated fluorinated carbon films. RSC Adv. 5(52), 41604–41607 (2015). https://doi.org/10.1039/C5RA06569K
He, X.Q., Kitipornchai, S., Liew, K.M.: Buckling analysis of multi-walled carbon nanotubes: a continuum model accounting for van der Waals interaction. J. Mech. Phys. Solids. 53(2), 303–326 (2005). https://doi.org/10.1016/j.jmps.2004.08.003
Feiler, A.A., Bergström, L., Rutland, M.W.: Superlubricity using repulsive van der Waals forces. Langmuir. 24(6), 2274–2276 (2008). https://doi.org/10.1021/la7036907
Yakubov, G.E., McColl, J., Bongaerts, J.H.H., Ramsden, J.J.: Viscous boundary lubrication of hydrophobic surfaces by mucin. Langmuir. 25(4), 2313–2321 (2009). https://doi.org/10.1021/la8018666
Thormann, E., Yun, S.H., Claesson, P.M., Linnros, J.: Amontonian friction induced by flexible surface features on microstructured silicon. ACS Appl. Mater. Interfaces. 3(9), 3432–3439 (2011). https://doi.org/10.1021/am200657d
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 51605336), the Foundation of the Department of Education of Guizhou province (Grant Nos. KY [2016] 009 and KY [2016] 106), the Key Research Program of Frontier Sciences, CAS (Grant Nos. QYZDY-SSW-JSC009 and U1737214), and Provincial Key Disciplines of Chemical Engineering and Technology in Guizhou Province (No. ZDXK[2017] 8).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, R., Zhao, J., Pu, J. et al. First-Principles Investigation on the Tribological Properties of h-BN Bilayer Under Variable Load. Tribol Lett 66, 124 (2018). https://doi.org/10.1007/s11249-018-1078-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11249-018-1078-y