
Abstract
Homozygous mice devoid of functional Prnp are resistant to scrapie and prion propagation, but heterozygous mice for Prnp disruption still suffer from prion disease and prion deposition. We have previously generated heterozygous cloned goats with one allele of Prnp functional disruption. To obtain goats with both alleles of Prnp be disrupted which would be resistant to scrapie completely, a second-round gene targeting was applied to disrupt the wild type allele of Prnp in the heterozygous goats. By second-round gene targeting, we successfully disrupted the wild type allele of Prnp in primary Prnp +/− goat skin fibroblasts and obtained a Prnp −/− cell line without Prnp expression. This is the first report on successful targeting modification in primary adult somatic cells of animals. These cells were used as nuclear donors for somatic cell cloning to produce Prnp −/− goats. A total of 57 morulae or blastocytes developed from the reconstructed embryos were transferred to 31 recipients, which produced 7 pregnancies at day 35. At 73 days of gestation, we obtained one cloned fetus with Prnp −/− genotype. Our research not only indicated that multiple genetic modifications could be accomplished by multi-round gene targeting in primary somatic cells, but also provided strong evidence that gene targeting in adult cells other than fetal cells could be applied to introduce precise genetic modifications in animals without destroying the embryos.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prion diseases, such as scrapie in goats or sheep and bovine spongiform encephalopathy (BSE) in cattle, are a group of fatal and infectious neurodegenerative disorders of the central nervous system (CNS) (Prusiner 1998). The therapeutic research for prion diseases is intensive but they are still currently incurable (Stewart et al. 2008). There is considerable evidence that the PrP plays an essential role in prion diseases (Aguzzi et al. 2008). More interestingly, while the cellular PrP (PrPC) is absolutely required for disease pathogenesis, it is dispensable for normal animal development. Mice devoid of functional Prnp (Prnp −/−) do not present macroscopic developmental or anatomical alternations, and they are completely resistant to scrapie and do not propagate prions (Büeler et al. 1992; Büeler et al. 1993). Also, PrP-deficient cattle are clinically, physiologically, histopathologically, immunologically and reproductively normal, the brain tissue homogenates from Prnp −/− cattle are resistant to prion propagation in vitro as assessed by protein misfolding cyclic amplification (PMCA) (Richt et al. 2007). Based on these results, goat, a natural host of the prototype of prion diseases scrapie, with PrP genes disruption should also survive and reproduce normally.
Different from Prnp −/− mice, the mice heterozygous for disrupted Prnp (Prnp +/−) still suffer from prion disease though with a prolonged incubation time, and the prion deposition and lesion distribution at terminal stage of disease are similar in heterozygous and wild type mice (Büeler et al. 1994; Manson et al. 1994). We have reported earlier on generation of five heterozygous cloned goats with one allele of Prnp be functionally disrupted (Yu et al. 2006). To obtain goats with both alleles of Prnp disruption which would be resistant to scrapie and prion propagation completely, the heterozygous goats could be bred to produce homozygosity. Alternatively, a second-round gene targeting could be carried out in Prnp +/− fibroblasts to disrupt the wile type allele of Prnp and followed with animal cloning.
Because livestock like goats, sheep, pigs, and cows have a long generation interval, the time required for production of homozygous livestock by sequential gene targeting could be greatly reduced compared to the traditional breeding strategy (Kuroiwa et al. 2004). In this research, while making efforts to produce homozygous goats by breeding traditionally, we also applied a second-round gene targeting in Prnp +/− fibroblasts to disrupt the wild type allele of Prnp, and then prepared cloned goats through nuclear transfer. Different from traditional somatic cell gene targeting in fetal fibroblasts (McCreath et al. 2000; Denning et al. 2001; Dai et al. 2002; Lai et al. 2002; Kuroiwa et al. 2004; Yu et al. 2006; Rogers et al. 2008), we chose the skin fibroblasts derived from an adult Prnp +/− goat ear biopsy as target cells for second-round gene targeting because Prnp +/− fetal is rare and difficult to obtain and the cloning efficiency of goat using fetal fibroblast or adult fibroblasts are almost the same in our laboratory.
In this report, we successfully targeted the wild type allele of Prnp in primary Prnp +/− skin fibroblasts and obtained a Prnp −/− cell line without Prnp expression. To our knowledge, it is the first report on successful targeting modification of a gene in primary adult somatic cells of animals. Using these targeted skin fibroblasts as nuclear donor in nuclear transfer, we finally obtained a cloned fetus with both alleles of Prnp be disrupted at day 73 of gestation.
Methods
Isolation and culture of Prnp +/− goat skin fibroblasts
The primary goat skin fibroblasts GSF3-1 were isolated from an ear skin biopsy of a 1.2-year-old male Prnp +/− Saanen dairy goat as described by Kubota et al. (2000). Briefly, skin biopsy was cut into small pieces after washing in PBS containing penicillin and streptomycin for 3 times and transferred into 100 mm tissue culture dishes containing 4 ml GMEM (Gibco) supplemented with 2 mM l-glutamine (Gibco), 1 mM sodium pyruvate (Gibco), 1× non-essential amino acids (Gibco), 10% FBS (Gibco), 100 U penicillin ml−1 and 100 μg streptomycin ml−1 (Gibco). When the expanding cells became confluent, they were disaggregated by 0.25% trypsin-EDTA (Gibco) and passaged to new dishes. The cells at different passages were cryopreserved in 10% DMSO (Sigma) for further manipulation.
Construction of the second-round targeting vector
Two fragments including the homologous arms around the exon 3 of Prnp in the targeted allele (Allele A) and the wild type allele (Allele B) of GSF3-1 were amplified, respectively. A 1.9 kb 5′ homologous arm and a 6.1 kb 3′ homologous arm from the two fragments of allele B were used to construct a promoter-less targeting vector GTPrPpuro with puro-pA sequence directly adjacent to the endogenous gene start codon. The 1.9 kb 5′ homologous arm was amplified by using primers PrP5f: 5′-GAACGTCGACTCTCCAGTCCATGGTCGTTCCTC-3′ with an artificial restriction-enzyme sites (underlined) at its 5′ end for molecular cloning and vector linearization (Sal I), and PrP5r: 5′-GTGGGCTTGTACTCGGTCATGATGACTTCTCTGCAAAAT-3′; with a 3′ tail (22 bp; in bold) within the Prnp locus and a 5′ tail complementary to the start of puro coding sequences (17 bp). The 0.9 kb puro-pA fragment was amplified by using primers puroF: 5′-ATTTTGCAGAGAAGTCATCATGACCGAGTACAAGCCCAC-3′, with a 5′ tail (22 bp, in bold) within the Prnp locus and complementary to the 5′ homologous arm, and puroR: 5′-CGCGGATCCGCGCCCCAGCTGGTTCTTTCC-3′, with a sites (underlined) at its 5′ end for molecular cloning (Bam H I). These two fragments were used to prime from each other to give a 2.8 kb product which was ligated to a 6.1 kb right arm amplified by using primers PrP3f: 5′-CGCGGATCCGGATCCTGGTTCTCTTTGTGG-3′ with a site for molecular cloning (Bam H I) and PrP3r: 5′-CCGCTCGAGGTCGACATGCTGGAGAGGATGTGGAGA-3′, with two sites for molecular cloning (Xho I) or vector linearization (Sal I) to complete the targeting vector.
The targeting vector GTPrPpuro was linearized with Sal I before electroporation.
Transfection and selection of the GSF3-1
The GTPrPpuro targeting vector was linearized with Sal I and introduced into passage 4 GSF3-1 by electroporation. About 1.0 × 107 exponentially growing GSF3-1 cells were disaggregated and washed in PBS twice, then mixed with 10 μg linearized and purified GTPrPpuro and subjected to a pulse of 400 V/250 μF in a 0.4 cm Gene Pulser Cuvette (Bio-Rad). The transfected cells were plated into two 10 cm-dishes in GMEM without selection. After 48 h, all cells were trypsinized and reseeded in selective cell-culture medium with 0.8 μg/ml Puromycin (Sigma). After 9–10 days selection, healthy and well-separated colonies were isolated with cloning rings and transferred to 48-well cell-culture plates. At subconfluence, half of cells were isolated for PCR analysis and the remaining cells were expanded by passaging until sufficient cells were obtained for cryopreservation and nuclear transfer.
Genomic PCR analysis of drug resistant cell colonies
Drug-resistant colonies were screened for targeting events by three different sets of PCR amplification across the 5′-homologous arm or the 3′-homologous arm. Approximately 5,000 cells in 48-well plates were lysed in 40 μl embryo lysis buffer (ELB) (40 mM Tris/HCl, pH 8.9, 0.9% Triton X-100, 0.9% Nonidet P-40, 0.4 mg/ml proteinase K) at 65°C for 15 min and heated to 95°C for 10 min to inactivate the proteinase K. PCR amplification was performed in a 20 μl reaction volume using the TaKaRa LA system with GC buffer with 3 μl cell lysate as DNA template. The positions of primers are indicated in Fig. 1. The primer sequences were: P1, 5′-CACAGCCAGGCATTCAGAAAC-3′; P2, 5′-AGTTGCCAGCCATCTGTTGTT-3′; P3, 5′-AACAACAGATGGCTGGCAACT-3′; P4, 5′-CACGATAGTAACGGTCCTCATAGTC-3′; P5, 5′-GCAGAGGACCCAAACAGACAT-3′; The thermal cycling conditions were: 5 min at 94°C; 30 cycles of 30 s at 94°C, 30 s at 62°C and 3.5 min (P1/P3 and P1/P4) or 6.5 min (P2/P5) at 72°C; followed by 10 min at 72°C.The PCR products of positive colonies were sent to sequencing to further confirm the targeting events.

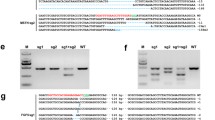
Diagrams of the targeted allele A, allele B, second-round targeting vector GTPrPpuro and targeted allele B. The black box represents the coding sequence (CDS) of Prnp, the open boxes represent neo-pA and puro-pA cassettes. PCR primers are indicated in the targeted Prnp locus and the RT-PCR primers are indicted in wild type Prnp locus. The expected size of PCR and RT-PCR products are also shown in the maps
RT-PCR analysis
Analysis of Prnp expression was performed in skin fibroblasts. Total RNA was extracted from Prnp +/+, Prnp +/− and Prnp −/− skin fibroblasts by using TRIzol reagent (Invitrogen), the first strand cDNA was synthesized with 2 μg RNA using RT-PCR kit (TaKaRa) following the manufacturer’s instructions. Subsequent PCR was carried out using primers P6 and P7 in 30 cycles of 94°C, 30 s, 58°C, 30 s, 72°C, 45 s. To detect expression of goat β-actin mRNA as control, primers GBAF and GBAR were used in the same PCR conditions. To exclude the possibility of genomic DNA contamination, another RT-PCR was carried out without reverse transcriptase. The positions of primers P6 and P7 are indicated in Fig. 1. The primers sequences were: P6, 5′-GAGTGCTGAAGAGTTGATGC-3′; P7, 5′-CTTACCAGTCCAACACTAGCA-3′; GBAF, 5′-TGGCACCACACCTTCTACAA-3′; GBAR, 5′-TCCTTGATGTCACGGACGAT-3′.
Western blot analysis
Total protein was extracted from Prnp +/+, Prnp +/−, and Prnp −/− skin fibroblasts with RIPA lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) at 4°C. Equal amounts of protein sample were run on a 12% SDS-PAGE gel and transferred to PVDF membranes by semi-dry electroblotting (Bio-Rad). After preincubation for 1 h in blocking buffer (25 mM Tris, pH 8.0, 140 mM NaCl, 3 mM KCl, 0.05% Tween-20, 5% non-fat dry milk), the membrane were incubated for 1 h in the same buffer containing a 1:2,000 dilution of a mouse anti-PrP monoclonal antibody 4C6 (National BSE Reference Laboratory, Qingdao, China) or a 1:1,000 dilution of a mouse anti-actin monoclonal antibody (Sigma, catalog no. A4700). After washing, the membranes were incubated for 1 h in the blocking buffer containing a 1:1,000 dilution of the horseradish peroxidase-conjugated goat anti-mouse IgG antibody. Then the membranes were washed again and visualized with diaminobenzidine (DAB).
Chromosome analysis
Targeted cells (Prnp −/−) were cultured in 100 mm dishes to about 80% confluence and arrested at metaphase (M) by adding colecmid (Sigma) to the culture at a final concentration of 0.5 μg/ml. After 1 h, the cells were collected and treated with hypotonic KCl (0.075 M) for 15 min at 37°C. The cells were then fixed in acetic methanol (vol/vol = 1:3), and drops of cell suspension were spread on clean microscopic slides. The chromosomes were stained with 5% Giemsa for 10 min. The numbers of well spread chromosomes within a clear cell boundary were counted under a light microscope at 1,000× magnification under oil.
Embryonic cloning
The cloned goat embryos were produced by nuclear transfer as described previously (Zou et al. 2002) with slight modification. In brief, the healthy targeted cells (Prnp −/−) with normal karyotypes were treated with starved medium and then introduced into the perivitelline of enucleated oocytes with a beveled pipette, subsequently, the reconstructed oocytes were electrically activated and cultured in oviducts of temporary recipients. The morulae and blastocytes from reconstructed embryos cultured in vivo for 5 days were surgically transferred into uteri of the synchronized final recipients. At day 35, the surrogates were scanned with a B-ultra-sound scanner to identify pregnant goats which were singled out and observed closely until they gave birth.
All animal work was done following a protocol approved by Shanghai Municipal Experimental Animal Committee.
Genomic PCR analysis of cloned embryo
Genomic DNA was extracted from the tissue of cloned goat embryo using FlexiGene DNA kit (QIAGEN) following the manufacturer’s instructions. PCR analysis was carried out using primers P1 and P4 to detect the genotypes of cloned embryo.
Results
Construction of the promoter-less targeting vector GTPrPpuro
The Prnp is expressed in goat skin fibroblasts, it is also possible to enrich for homologous recombination events using a promoter-trap strategy as in fetal fibroblasts.
In attempting to target the allele B, there is the possibility that the second targeting vector will undergo homologous recombination with the first integrated targeting vector GTPrP (Yu et al. 2006), resulting in replacement of the knockout vector in allele A rather than disruption of the allele B. Two fragments including the 5′ and 3′ homologous arm of the first targeting vector GTPrP were amplified from Prnp allele A of GSF3-1, the analogous fragments were also amplified from allele B. These fragments were analyzed and compared. The results indicated that the allele A and allele B of Prnp have 2% discrepancies in the homologous arms. To targeting the allele B as we expected, the targeting vector GTPrPpuro in which the homologous arms were isogenic to allele B was constructed by inserting the puro-pA directly adjacent to the initiation codon of Prnp. If homologous recombination occurs between GTPrPpuro vector and allele B, a 23 bp coding region followed the initiation codon will be deleted and replaced by the 0.9 kb puro-pA cassette (Fig. 1). But if the recombination occurs in allele A, the fist targeting vector GTPrP will be replaced by the second-round targeting vector GTPrPpuro. The same as the first vector GTPrP, the GtPrPpuro also retained the splice acceptor site of exon 3 to avoid causing severe ataxia and Purkinje cell loss in aged animal (Moore et al. 1999; Rossi et al. 2001).
Targeting of the allele B of Prnp with GTPrPpuro vector
Linearized GTPrPpuro vector was delivered into passage 4 GSF3-1 fibroblasts by electroporation. After Puromycin selection for 9–10 days, the drug-resistant colonies were isolated using cloning ring. Puromycin-resistant colonies were first screened by PCR using P1/P4 to detect targeted events. Of 204 colonies analyzed by PCR using a forward primer, P1, that is located upstream of the 5′ homologous arm and a reverse primer, P4, that is located within the 3′ homologous arm, five colonies (2.5%) were found to have targeted allele B to produce cells with both alleles of Prnp be disrupted (Prnp −/−), as determined by the presence of two bands of the expected sizes: a 3.5 kb band from the first targeted allele A and a 3.7 kb band from the second targeted allele B (Fig. 2a). Other five colonies (2.5%) were found to have replaced the first targeting vector in allele A as determined by the presence of two bands: a 2.8 kb band from the normal Prnp locus in allele B and a 3.7 kb band from the replaced allele A (Fig. 2a; Table 1). These results indicated that the vector GTPrPpuro had no bias on targeting efficiency for allele A and allele B of Prnp though it was isogenic to allele B.

PCR analysis of Puromycin-resistant colonies. Colony numbers are indicated above each lane. The PCR primers are (a) P1/P4, (b) P1/P3, (c) P2/P5. The position of primers are indicated in Fig. 1. No. 19 and No. 20 colonies were Prnp −/− colonies as the presence of a 3.5 kb and a 3.7 kb bands amplified by P1/P4, a 3.0 kb band amplified by P1/P3, and a 6.5 kb band amplified by P2/P5. No. 21, No. 22 colonies were untargeted colonies. No. 23 colony was replaced colony as the presence of a 2.8 kb and a 3.7 kb bands amplified by P1/P4. GSF3-1 was untransfected goat skin fibroblast. M: 1 kb DNA ladder
To further confirm the successful targeting events, two additional independent PCRs were carried out in the targeted colonies. The results were as expected (Fig. 2b, c). In addition, the 3.0 kb PCR products generated with P1/P3 and the 6.5 kb PCR products generated with P2/P5 from positive colonies were sequenced and the results were also consistent with our expectation (Fig. 3). These results firmly indicated the targeting events by homologous recombination had been occurred between the targeting vector GTPrPpuro and Prnp locus in GSF3-1. We successfully targeted a gene in adult somatic cells and obtained a cell line with both alleles of Prnp disruption.

Sequencing analysis of the targeted allele. (a) The sequences of the connection between the 5′ homologous arm of GTPrPpuro and Prnp locus. (b) The sequences of the connection between the 3′ homologous arm of GTPrPpuro and Prnp locus. The results indicated successful homologous recombination between the targeting vector GTPrPpuro and Prnp locus
Functional disruption of Prnp expression in skin fibroblasts
To evaluate the functional disruption of Prnp gene in fibroblasts, RT-PCR was carried out to detect the mRNA level of Prnp in Prnp +/+, Prnp +/−, and Prnp −/− goat skin fibroblasts. Primers P6 and P7 are located in the exon 3 of Prnp. Amplification with P6 and P7 generated a 446 bp fragment indicated the expression of the wild type allele of Prnp in Prnp +/+ and Prnp +/− fibroblasts. No fragment was generated in Prnp −/− fibroblasts in which both alleles of Prnp had been disrupted by insertion of neo-pA and puro-pA cassette (Fig. 4). These results indicted that the transcription of Prnp had been functionally disrupted in skin fibroblasts.

RT-PCR analysis of Prnp +/+, Prnp +/−, and Prnp −/− goat skin fibroblasts. By the presence or absence of expected size of RT-PCR products, Prnp transcription were observed in Prnp +/+ and Prnp +/− fibroblasts but not in Prnp −/− goat skin fibroblast with both alleles be disrupted. β-actin was used for control to monitor template amounts
Western blot was also performed on Prnp +/+, Prnp +/−, and Prnp −/− goat skin fibroblasts using an anti-PrP monoclonal antibody 4C6 to confirm the functional inactivation of Prnp. PrP-specific bands (approximately 35 kDa) were detected from both Prnp +/+ and Prnp +/− skin fibroblasts, but no band was observed in Prnp −/− skin fibroblasts (Fig. 5). These results clearly demonstrated that the expression of Prnp was functionally disrupted in goat skin fibroblasts.

Western blot analysis of Prnp +/+, Prnp +/−, and Prnp −/− goat skin fibroblasts. The absence of PrP-specific band in Prnp −/− goat skin fibroblasts indicated the functional disruption of Prnp expression in goat skin fibroblasts. The anti-actin antibody was used to ensure that each lane contained an equal amount of total protein
Production of Prnp −/− embryo by nuclear transfer
The healthy No. 19 targeted skin fibroblasts (Prnp −/−) with normal karyotype (data not shown) were used as nuclear donor for reconstructing embryos with enucleated oocytes. After nuclear transfer, a total of 57 (40.1%) morulae or blastocytes were transferred to 31 recipients, which produced 7 (22.6%) pregnancies at day 35. But unfortunately, all pregnancies aborted subsequently with only 1 of the fetuses recovered at day 73 of gestation (Table 2). Genomic DNA analysis by PCR using primers P1 and P4 was carried out to detect the genotype of this cloned embryo, the results show that this aborted fetus was Prnp −/−, which was consistent with the donor cell No. 19 colony (Fig. 6).

Genomic PCR analysis of the aborted cloned fetus. 1: Wild type skin fibroblasts (Prnp +/+); 2: Heterozygous skin fibroblasts GSF3-1 (Prnp +/−); 3: No.19 skin fibroblasts colony with allele B targeted (Prnp −/−); 4: No.23 skin fibroblasts colony with allele A replaced (Prnp +/−); 5: Aborted cloned fetus. M: 1 kb DNA ladder
Discussion
By second-round gene targeting, we successfully targeted the wild type allele of Prnp in primary Prnp +/− skin fibroblasts using a promoter-less vector GTPrPpuro. The targeting modification in adult somatic cells may have wider application, because the adult skin fibroblasts are more convenient than fetal fibroblasts to obtain and have no ethical issues. In contrast to the gene targeting in adult rhesus macaque fibroblasts (Meehan et al. 2008), the primary adult goat fibroblasts without transfection of hTERT expression cassette could be cultured in vitro long enough to complete gene targeting procedures, and the homologous recombination could be successfully achieved without S-phase synchronization or adding SV 40 enhancer element to the targeting vector in our experiments. Our research also indicated that multiple genetic modifications could be accomplished by multi-round gene targeting in primary adult somatic cells.
To specifically target the allele B of Prnp, we constructed a second targeting vector in which the homologous arms were isogenic to allele B, which has 2% discrepancies to allele A in this location. But the results indicated that there were no bias on the targeting efficiency between the allele A and allele B of Prnp (1:1) using this vector. Our data, together with others (Sedivy et al. 1999; Kuroiwa et al. 2004), supported a postulation that the isogenicity does not significantly influence the efficiency of homologous recombination in some gene locus in goat skin fibroblast.
Although we have not obtained live-born Prnp −/− goats by second-round gene targeting and nuclear transfer, we did not expect this failure as a direct result of the gene disruption of Prnp because three Prnp −/− goats obtained through breeding strategy remained healthy up to 5 months of age (data not shown). The aborted cloned fetus had similar abnormalities as other nuclear transfer experiments with untransfected cells in our laboratory. And the development rates at different stages of the animal cloning procedure were also similar with other experiments (Yu et al. 2006). So we attributed this result to the inherently low efficiency of somatic cell cloning technology (Wilmut et al. 1997).
All the Prnp −/− goats obtained from breeding strategy have remained healthy for at least 5 months without showing obvious abnormalities, this indicated that “loss of function” of goat PrPc is unlikely in itself to be significant in the pathogenesis of scrapie and that ablation of the normal cellular prion protein PrPc function does not adversely affect the normal goat development. Therefore, our research supplied more evidence supporting a general hypothesis that PrPc function is not vital for normal animal development (Tremblay et al. 1998; Mallucci et al. 2002). However, detailed characterization of these goats should be carried out to investigate the physiological function of PrPc in goats.
The Prnp −/− goats could be a more relevant model than PrP-deficient mice for elucidating the basic mechanisms of prion diseases and PrPc functions. They also could be a better source of a variety of goat-derived products that have been extensively used in biotechnology, and be useful for production of prion-free therapeutic recombinant human protein, tissue and organs in transgenic goats for biomedical application.
Abbreviations
- DMSO:
-
Dimethyl sulfoxide
- Dpl:
-
Doppel protein
- FBS:
-
Fetal bovine serum
- GMEM:
-
Glasgow minimal essential medium
- neo :
-
Neomycin phosphotransferase gene
- Prnp :
-
Prion protein gene
- PrP:
-
Prion protein
- puro :
-
Puromycin N-acetyl-transferase gene
References
Aguzzi A, Sigurdson C, Heikenwaelder M (2008) Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 3:11–40. doi:10.1146/annurev.pathmechdis.3.121806.154326
Büeler H, Fischer M, Lang Y et al (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356(6370):577–582. doi:10.1038/356577a0
Büeler H, Aguzzi A, Sailer A et al (1993) Mice devoid of PrP are resistant to scrapie. Cell 73(7):1339–1347. doi:10.1016/0092-8674(93)90360-3
Büeler H, Raeber A, Sailer A et al (1994) High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med 1(1):19–30
Dai Y, Vaught TD, Boone J et al (2002) Targeted disruption of the a1,3-galactosyltransferase gene in cloned pigs. Nat Biotechnol 20(3):251–255. doi:10.1038/nbt0302-251
Denning C, Burl S, Ainslie A et al (2001) Deletion of the a(1,3)galactosyl transferase (GGTA1) gene and the prion protein (PrP) gene in sheep. Nat Biotechnol 19(6):559–562. doi:10.1038/89313
Kubota C, Yamakuchi H, Todoroki J et al (2000) Six cloned calves produced from adult fibroblast cells after long-term culture. Proc Natl Acad Sci USA 97(3):990–995. doi:10.1073/pnas.97.3.990
Kuroiwa Y, Kasinathan P, Matsushita H et al (2004) Sequential targeting of the genes encoding immunoglobulin-mu and prion protein in cattle. Nat Genet 36(7):775–780. doi:10.1038/ng1373
Lai L, Kolber-Simonds D, Park KW et al (2002) Production of a-1,3-galactosyltransferase knockout pigs by nuclear transfer cloning. Science 295(5557):1089–1092. doi:10.1126/science.1068228
Mallucci GR, Ratté S, Asante EA et al (2002) Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 21(3):202–210. doi:10.1093/emboj/21.3.202
Manson JC, Clarke AR, McBride PA et al (1994) PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 3(4):331–340
McCreath KJ, Howcroft J, Campbell KH et al (2000) Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 405(6790):1066–1069. doi:10.1038/35016604
Meehan DT, Zink MA, Mahlen M et al (2008) Gene targeting in adult rhesus macaque fibroblasts. BMC Biotechnol 8:31. doi:10.1186/1472-6750-8-31
Moore RC, Lee IY, Silverman GL et al (1999) Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 292(4):797–817. doi:10.1006/jmbi.1999.3108
Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95(23):13363–13383. doi:10.1073/pnas.95.23.13363
Richt JA, Kasinathan P, Hamir AN et al (2007) Production of cattle lacking prion protein. Nat Biotechnol 25(1):132–138. doi:10.1038/nbt1271
Rogers CS, Hao Y, Rokhlina T et al (2008) Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 118(4):1571–1577. doi:10.1172/JCI34773
Rossi D, Cozzio A, Flechsig E et al (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J 20(4):694–702. doi:10.1093/emboj/20.4.694
Sedivy JM, Vogelstein B, Liber HL et al (1999) Gene targeting in human cells without isogenic DNA. Science 283(1):9. doi:10.1126/science.283.5398.9a
Stewart LA, Rydzewska LH, Keogh GF et al (2008) Systematic review of therapeutic interventions in human prion disease. Neurology 70(15):1272–1281. doi:10.1212/01.wnl.0000308955.25760.c2
Tremblay P, Meiner Z, Galou M et al (1998) Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc Natl Acad Sci USA 95(21):12580–12585. doi:10.1073/pnas.95.21.12580
Wilmut I, Schnieke AE, McWhir J et al (1997) Viable offspring derived from fetal and adult mammalian cells. Nature 385(6619):810–813. doi:10.1038/385810a0
Yu G, Chen J, Yu H et al (2006) Functional disruption of the prion protein gene in cloned goats. J Gen Virol 87(Pt4):1019–1027. doi:10.1099/vir.0.81384-0
Zou X, Wang Y, Cheng Y et al (2002) Generation of cloned goats (Capra hircus) from transfected foetal fibroblast cells, the effect of donor cell cycle. Mol Reprod Dev 61(2):164–172. doi:10.1002/mrd.1143
Acknowledgments
We gratefully thank Hongying sha, Laixiang Ge, Dawei Hu, Fazhan Xie and Lijun Ding for their technical assistance to this work. This work was supported by grants from Shanghai Rising-Star Program (07QB14022) and China Postdoctoral Science Foundation (20060400174).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhu, C., Li, B., Yu, G. et al. Production of Prnp −/− goats by gene targeting in adult fibroblasts. Transgenic Res 18, 163–171 (2009). https://doi.org/10.1007/s11248-008-9220-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-008-9220-5