
Abstract
Plasmas and gas discharges in contact with liquids have played an important role in the history of chemical processing and scientific inquiry, leading to the discoveries of elements such as argon and compounds such as ozone. Recently-developed atmospheric-pressure plasma sources have renewed the study of plasma–liquid systems with applications in chemical processing, materials synthesis, and chemical analysis. In many cases, these approaches utilize glow discharge electrolysis configurations where a DC plasma replaces one of the metal electrodes in a standard electrolytic cell. These configurations have been used to great effect for the synthesis of various nanomaterials and more recently, in the processing of carbon dioxide. In this work, we overview recent developments using plasmas as electrodes in electrolytic cells for chemical processing, drawing parallels to conventional electrochemistry and electrocatalysis. In particular, we highlight recent studies on the fundamental chemical processes at the plasma–liquid interface, including new interfacial measurement techniques used to probe charge transfer. We conclude with an overview of opportunities for these configurations in the future and highlight the need for further fundamental study.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Historically, electrochemistry has played an important role across a number of technologies, whether it be the electrolytic cell for electrolytic processing or the galvanic cell for energy production. In the conventional electrochemical cell, reduction and oxidation (redox) reactions are mediated at the interface between solid electrodes and an electrolyte fluid. For this reason, electrocatalysts are typically utilized to promote reactions at the electrode surface. As such, there has been a tremendous amount of research in the catalytic behaviour of electrode materials and catalyst development for applications such as carbon dioxide (CO2) processing [1], water electrolysis [2], and fuel cells [3].
An alternative approach to electrochemical catalysis is to replace one or both of the solid electrodes with a plasma (also called a gas discharge). In this case, one or both of the reduction and oxidation reactions occur at the interface of the plasma and the electrolyte. Plasmas are incredibly reactive environments, where a neutral gas is broken down into a chemical cocktail of free ions, free electrons, molecular and atomic radicals, excited metastables, and photons (Fig. 1a). These species themselves, as well as secondary species produced at the vapor interface and in the solution, can drive electrochemical reactions. Plasma electrochemistry stands in stark contrast to conventional electrocatalysis. Rather than the properties of the solid state electrocatalyst driving the electrochemical process, it is the properties of free radicals produced in the plasma that are important. In essence, plasma electrochemical systems offer a “catalyst-free” approach to electrochemistry.

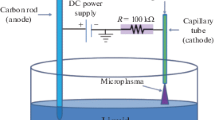
a Schematic of the plasma–liquid interface and the reactive species that dissolve from the plasma phase into the liquid phase, inducing many reactions. b Schematics of typical DC plasma–liquid configurations as an electrolytic cell (left). The plasma anode (liquid cathode) configuration (right). The plasma cathode (liquid anode) configuration
Although plasma–liquid experiments can be traced back for centuries, over the past two decades, emerging approaches to plasma generation have opened up new avenues for plasma–liquid processing. Recently, techniques based on plasmas “in” and “in contact with” liquids have rapidly grown as the transfer of chemical species across the gas/liquid interface opens up new opportunities for chemical processing. In this paper, we overview recent advances in the areas of chemical processing and chemical analysis as well as understanding the complex interface between a plasma and liquid. In particular, we discuss recent work by the authors’ group aimed at deciphering the fundamental interfacial processes that occur, focusing on efforts to understand electron transfer reactions. Such fundamental advances will play a critical role as plasma–liquid technologies continue to develop and new opportunities for catalyst-free electrochemistry emerge. For the purposes of brevity, we will focus on plasmas “in contact with” liquids, often called glow discharge electrolysis. We leave out any extensive discussion on plasmas “in” liquids, often called contact glow discharge electrolysis, and point the reader to other works, such as the review by Gupta [4].
2 Background
2.1 History of Plasma–Liquid Studies
Plasmas in contact with liquids have played an important role in scientific discovery and technology for centuries. Dating to at least the late 1700s with the work of Cavendish, and including the likes of Priestly, Rayleigh, and Ramsay, arcs generated over water have been used to understand the composition of air [5] and have also played a critical role in the discoveries of argon (Ar) [6] and ozone (O3) [7]. They also have a long history in chemical processing, and in the late 1800s, Birkeland and Eyde used arcs over water for the industrial production of nitrogen-based fertilizers [8]. Similarly, the likes of Gubkin [9] and Klemenc [10] among others in the later 19th and early twentieth centuries used plasmas to produce colloidal silver (Ag) particles and hydrogen peroxide (H2O2), respectively.
While there are many configurations that can be used for bringing the reactive species from a plasma into a liquid [11], of prime interest in this work is conditions where the liquid itself serves as a conductive element in a direct current (DC) plasma circuit (Fig. 1b). Typically, one electrode is submerged and the other is suspended over the solution, rather than being submerged. By applying a high voltage between the electrodes (~kV), the interstitial gas between the liquid and the suspended electrode breaks down to form a conductive plasma.
This simple configuration is analogous to two standard circuits. From a plasma perspective, it represents the classic glow discharge configuration, which typically consists of two electrodes operated under DC conditions in a vacuum tube, and is a non-equilibrium plasma, which means that the gas and ion temperatures are much lower than the electron temperature. While sustaining a glow discharge at atmospheric pressure is ostensibly difficult as gas heating leads the plasma to become thermal, recent strategies such as miniaturization (so-called microplasmas) have overcome these limitations [12]. Using a liquid as the counter-electrode is particularly useful because its high heat capacity helps keep the plasma relatively cool. From the liquid perspective, the plasma configuration resembles that of an electrolytic cell where one of the electrodes has been replaced by a plasma. For this reason, this field is often referred to as plasma electrochemistry. Depending on the orientation, the plasma is the anode in the electrolytic cell as shown in Fig. 1b, left (that is, the liquid is the cathode to the plasma) or the plasma serves as the cathode as shown in Fig. 1b, right (with liquid acting as the anode to the plasma).
These types of plasma–liquid configurations were studied extensively by Hickling and his co-workers over a nearly 25 year period in the mid-twentieth century (largely summarized in [13]). Typically, the studies focused on comparing plasma electrochemistry, which they termed ‘glow discharge electrolysis’, to conventional electrochemistry. Among their many findings, of particular note was that the oxidation rate of a plasma anode was greater than the maximum allowed by Faraday’s law for a metallic anode. This is because the positive ions incident on the liquid surface carry enough energy to break one or more water molecules into H· and OH· or (e−)aq and H2O+ radicals, with OH· and H2O+ being powerful oxidizing agents [14].
2.2 Developments in Plasma Electrochemistry Applications
In 1993, Cserfalvi et al. showed the first practical use of glow discharge electrolysis by employing it as a tool for chemical analysis. They used the plasma anode configuration (Fig. 1b, left) and conducted atomic optical emission spectroscopy (OES) of the plasma above the liquid surface [15]. They proposed that the plasma effectively sputtered elemental components dissolved in the liquid into the gas phase, where they could be excited by the plasma and detected by their optical emission. While the exact mechanisms are still being investigated, their work helped usher in this approach to atomic emission spectroscopy as a potentially cheap, rapid, and simple alternative to conventional inductively coupled plasma (ICP) OES for elemental analysis in applications such as water testing and process monitoring.
Since that time, a number of advances have been made in both the experimental configuration as well as quantitative performance of plasma electrochemistry-based OES techniques. A variety of configurations have been developed to facilitate both sample introduction and optical access, including direct sample injection, jets, and sprays, and reviews by Mezei and Cserfalvi [16], Webb and Hiefjte [17], and Jamorz et al. [18] have summarized these advances. Impressively, a wide variety of analytical targets have been demonstrated and quantified, as summarized in [18], and methods for introducing standards have been developed [19]. Importantly, all of this progress has recently led to the commercialization of this technique, with products now beginning to appear in the market place based on plasma electrolysis OES (e.g., the MH-5000 Elemental Analyzer by Micro Emission Ltd., Ishikawa, Japan).
While the plasma cathode configuration has not been extensively explored studied for chemical analysis, it has become a popular technique for material synthesis. The first demonstration of this was the plasma electrochemical analogue of electroplating. Unlike, an electrolytic cell, however, there is no solid electrode for the reduced cations to adsorb on, and thus the neutral metal atoms agglomerate to form colloidal nanoparticles as shown in Fig. 2. Sankaran et al. demonstrated this using silver (Ag+) and gold (Au3+) cations [20]. Using either a metal anode (as shown in Fig. 3) or an aqueous solution of silver nitrate (AgNO3) or chloroauric acid (HAuCl4), they were able to produce Ag and Au nanoparticles with mean sizes ~5–10 nm. They argued that plasma-injected electrons directly reduce the metal cations, analogous to direct electron transfer to a cation absorbed on a metal electrode.

Courtesy of R. Mohan Sankaran, Case Western Reserve University
Schematic of colloidal nanoparticle synthesis using a liquid anode (plasma cathode) configuration.

Schematic of proposed charge transfer processes in a liquid cathode (plasma anode) and b liquid anode (plasma cathode) configurations. In the former, the water is ionized by bombarding plasma ions to form solvated electrons—a process termed the modified Hart-Anbar cycle [45]. In the latter, the plasma electrons directly solvate before reacting away
However, multiple studies have suggested that direct reduction by plasma electrons is potentially not responsible for Au nanoparticle synthesis from solutions of HAuCl4 [21, 22]. For example, Patel et al. first exposed a solution without any HAuCl4 to a plasma and added the HAuCl4 after shutting the plasma off [22]. Since Au nanoparticles were still synthesized, this strongly suggested that the reducing species generated by the plasma was long-lived, whereas injected electrons at any significant concentration would be short-lived. Similarly, they used an H2O2 scavenger, titanium oxysulfate (TiOSO4), to inhibit nanoparticle growth suggesting the plasma-generated H2O2 was causing the growth. In fact, in a novel approach they call dual plasma electrolysis, Shirai et al. showed they could use an H-cell electrochemical configuration with plasmas as both the anode and cathode simultaneously. They were able to synthesize both Ag and Au nanoparticles in both the cathodic and anodic cells, indicating that critical reducing species can be generated in either bias configuration, even though plasma electrons are ostensibly only injected in the cathodic cell [23]. Interestingly, they showed they could also form Au/Ag core/shell nanoparticles when using solutions of both AgNO3 and HAuCl4.
In the past several years, the field of plasma-based electrolytic synthesis has grown significantly with a number of configurations (aqueous and ionic liquids) and materials (metallic and non-metallic) being studied. For the most part, primarily metal nanoparticles have been synthesized including copper (Cu) [24–26], palladium (Pd) [27], and iridium (Ir) [28] in addition to Ag and Au. However, it has also been shown that if ionic liquids are used, both silicon (Si) and germanium (Ge) nanoparticles can be formed [29]. Impressively, Ghosh et al. have recently demonstrated a flowing liquid approach suggesting the imminent scalability of the process [30]. In addition to synthesis, it has also recently been shown that the surface modification of silicon (Si) NPs and exfoliation of graphene oxide (GO) are possible [31, 32], broadening the potential use of plasma electrochemistry for other forms of materials preparation. Overall, a recent review by Chen et al. summarizes the significant advances in materials processing by plasmas in contact with liquids [33].
2.3 Fundamental Plasma and Liquid Characterization
It is clear that the chemical and physical processes that occur at the plasma–liquid interface are extremely complex. For as long as glow discharge electrolysis has been used as a chemical tool, there have been studies trying to understand the chemistry and physics in these systems. On the plasma side, there has been an abundance of studies looking to characterize not only critical plasma parameters (such as the electron density, electron temperature, and gas temperature) but to identify important plasma species. The most frequently used technique is OES of the plasma, which target transitions that can be used to understand the plasma itself. This technique, for example, has been used to show that the hydroxyl radical (OH·) is produced in the interfacial vapour region above the plasma. Further, hydrogen (H) and nitrogen (N2) transitions, along with current–voltage characteristics, have been used to determine gas and electron temperatures and electron densities. Many of these advances have been summarized in recent reviews [11, 18, 34].
On the liquid side, the challenge to understand fundamental processes is complicated because many of the reactions consist of short-lived intermediates. For this reason, the general strategy has been to look at long-lived products of reactions driven either electrolytically or by other dissolved reactive plasma species. While the products of chemical processing mentioned above (e.g., nanoparticles) are good examples of the type of studies that could be conducted, a number of recent efforts have focused particularly on understanding the dissolution of reactive plasma species for applications in biology, water treatment, and surface processing [35, 36]. In particular, reactive oxygen and reactive nitrogen species (ROS and RNS, respectively) play a major role in plasma-based water treatment [37, 38] and plasma-based medicine [39]. A concise overview of the various bactericidal ROS and RNS produced by plasma can be found in Ref [39]. Species such as superoxide (\({\text{O}}_2^ -\)) and the hydroxyl radical (OH·) can be produced in the vapour layer at the plasma–liquid interface and dissolve into solution. In the liquid phase, it is assumed that most of the ROS follow various reaction paths that ultimately results in OH− and H2O2. Similarly, many of the RNS follow various reaction paths that ultimately lead to nitrous acid (HNO2) and nitric acid (HNO3).
While optical techniques have been used with great success for measuring radicals in the plasma phase, such measurements are far more elusive in the liquid phase, where the mean-free-path and overall lifetime of radicals is much shorter. One recent advance for understanding these complex systems is the development of sophisticated modelling tools. Simulations of atmospheric pressure plasmas in contact with solutions can now capture the electromagnetic field and reactive transport of various radicals in both the plasma and liquid phases [40–43]. Using experimentally measured rate constants and diffusivities, these simulations can predict which radicals will be most abundant in the liquid phase. Recently, molecular dynamic simulations by Yusupov et al. provided an atomistic picture of how O· and OH· radicals from the plasma phase become solvated [44]. Overall, simulations can provide important information about the liquid phase chemistry, filling in details where experimental methods come up short.
3 Recent Advances in Understanding Charge Transfer at the Interface
The early work on Ag and Au nanoparticle synthesis raised some interesting questions on the fundamental electrochemical mechanisms that occur in plasma cathode electrolytic cells. The work by Sankaran and his colleagues suggested that when the plasma was the cathode, electron transfer from the gaseous plasma phase to the liquid phase directly reduced metal cations [20]. Yet, as noted earlier, others made arguments that other plasma-produced reactive species, namely H2O2, are responsible for the reduction of the metal cations, at least in the case of Au [21, 22]. For this reason, the authors’ group and collaborators have conducted a variety of studies in order to understand the fundamental charge transfer process across the plasma–liquid interface, specifically focusing on the fate of plasma electrons.
Understanding how charge is transferred between the plasma and liquid phases is interesting not only from the perspective of the chemical process (redox reaction), but because it is also essential to complete the circuit and sustain the plasma. Classically, electrically-driven DC discharges are maintained by two processes—electron-impact ionization in the volume of the gas and secondary electron emission from the cathode surface into the plasma. For a plasma to be sustained with a liquid cathode (plasma anode), a similar emission process must occur—that is, electrons must be ejected from the solution into the gas phase. Alternatively, for a plasma acting as a cathode with a liquid anode, the electrons are injected into the liquid directly.
3.1 Plasma Anode Electron Transfer
For plasma anodes, Cserfalvi and Mezei proposed an emission mechanism that they call the modified Hart-Anbar process [16, 45], building upon earlier hypotheses from glow discharge electrolysis [46]. In this model, outlined in Fig. 3a, the positive discharge ions, mostly (H2O+)g formed in the vapor layer, but also air ions such as N2 + and O2 +, bombard the surface of the liquid and have sufficient energy to collisionally ionize the water molecules to produce (H2O+)aq and a solvated electron (e−)aq. These solvated electrons recombine with (H+)aq cations to produce a hydrogen radical H·. According to their hypothesis, these radicals diffuse into the gas phase to form (H·)g, where they are ionized by electron impact ionization in the discharge. This produces a free electron in the discharge phase (e−)g that originated from (H·)aq, and the resulting (H+)g dissolves back into solution to complete the cycle. Reduction reactions at the submerged cathode in the solution then serve the role of ensuring electrons are replenished in the solution and that current is conserved. Importantly, this process depends on an excess of (H+)aq in the solution and is thus pH dependent. Cserfalvi and Mezei showed that their proposed mechanism agrees with their observed relationship between the secondary emission coefficient and the pH of the solution cathode [45].
Because of this early evidence, in the analytical chemistry community it is now almost standard to conduct plasma electrochemistry-based OES at low pH (<2) conditions [17, 18], but it is not universally required. In fact, there are instances where basic solutions produce strong OES signals [47], and some configurations can operate at many pH conditions as long as the solution is sufficiently conductive [48]. Furthermore, it has also been observed that liquid is physically ejected from the liquid cathode surface via electrospray atomization, which also transfers species from liquid to gas [49]. This highlights that while the modified Hart-Anbar process may be necessary to sustain the plasma in some plasma anode configurations, other configurations rely on different mechanisms. In fact, Marcus and collaborators suggest that the high power density of the discharge (~10 W mm−3) in their configuration evaporates the solution, whose species are subsequently ionized, replenishing the electron supply in the discharge [50]. It is certainly the case that there is no current scientific consensus on the exact charge exchange mechanism in these systems, and as noted by Jamorz et al. [18], no measurements of discharge properties provide definitive evidence.
3.2 Plasma Cathode Electron Transfer
The focus of our work has been on the electron transfer in plasma cathode configurations, which is substantially different than a plasma anode as shown in Fig. 3b. In particular, electrons are initially injected into the plasma from the suspended metal electrode (Fig. 1b) rather than from the liquid, such that there is no need for electrons or ions to be ejected from the liquid to sustain the plasma. In this case, in order to complete the circuit, either free electrons or some other negatively-charged species in the plasma must be directly injected into the solution. Much of our work has focused on resolving this issue and determining the ultimate chemical paths of the injected species. The primary driver has been exploring the assertion that plasma electrons are directly injected into the solution where they solvate and drive electrochemistry.
Initially, studies on electron-driven chemistry at the plasma–liquid interface focused on effects that would persist for long times and ultimately change the bulk properties of the solution. One of the first of these studies was conducted by Sankaran and collaborators, who studied the classic ferri/ferrocyanide redox couple [51]. Cleverly, they used an H-cell configuration in order to isolate reduction reactions at the plasma cathode from oxidation reactions at the submerged metal (platinum) anode. Ferricyanide, \({\text{Fe}}\left( {{\text{CN}}} \right)_{\text{6}}^{{\text{3}} - },\) is well known to undergo a single electron reduction to form ferrocyanide, \({\text{Fe}}\left( {{\text{CN}}} \right)_6^{4 - }.\) Because \({\text{Fe}}\left( {{\text{CN}}} \right)_6^{3 - }\) absorbs strongly in the blue at ~420 nm, the colour of the solution gradually turns from yellow to transparent as the \({\text{Fe}}\left( {{\text{CN}}} \right)_6^{3 - }\) is reduced. By quantifying the absorbance using ultraviolet–visible (UV–Vis) spectroscopy, Sankaran and his group were able to show that indeed \({\text{Fe}}\left( {{\text{CN}}} \right)_6^{3 - }\) was being reduced to \({\text{Fe}}\left( {{\text{CN}}} \right)_6^{4 - }\) as shown in Fig. 4a, suggesting that it was plasma electrons that were directly responsible for the reduction reaction. Further, as shown in Fig. 4b, the percent of ferricyanide reduced increased linearly in time, suggesting the reaction proceeds faradaically. However, when they extracted the faradaic efficiency of the process, they observed it was <10% efficient. In order to attempt to account for the electrons not contributing to ferricyanide reduction, they used Ag and Cu anodes to measure the anodic dissolution due to oxidation reactions. These were in excellent agreement with Faraday’s law, “that charge is transferred with 100% efficiency from the plasma to the liquid” [51]. Yet, it was unclear where or how charge was being transferred for the electrons that were not reducing ferricyanide.

Adapted with permission from [51]. Copyright (2011) American Chemical Society
a UV–Vis absorbance spectra for solutions of ferricyanide after 0, 5, and 15 min of exposure to a 6 mA DC plasma. The reduction in the peak height at ~420 nm is confirmation of electrochemical reduction of ferricyanide to ferrocyanide by the plasma. b Percentage of the ferricyanide reduced by the plasma as a function of time for DC plasma currents of 3 and 6 mA.
One obvious cause for the low efficiency is that a large percentage of the electrons injected are consumed by water electrolysis. Plasma-injected electrons will become solvated before reacting with solution species. Furthermore, the electrons have a free energy of solvation of −156 kJ mol−1, so the solvated state is energetically favourable to the plasma state [52]. Once solvated, it is well known that solvated electrons are extremely reactive, with a reduction potential of −2.8 V, and can react through multiple pathways [52]. They typically undergo one of two processes. Either, they react with any dissolved scavenger S (anion, cation, or netural species) directly via
or they react directly with the water in a process called second-order recombination (essentially water electrolysis) via
While the ferricyanide is a scavenger in (1), Reaction (2) proceeds quite rapidly with a reaction rate constant of 2k e = 1.1 × 1010 M−1 s−1, accounting for the inefficiency observed in [51].
Our group, in collaboration with Sankaran’s group, explored this by directly measuring the products of Reaction (2), namely (H2)g and OH− [53]. Using mass spectrometry, we were able to measure (H2)g produced in the headspace of the reactor, confirming the hydrolytic process. Further, we could also detect the excess OH− produced by Reaction (2) because it leaves the solution locally basic beneath the plasma–liquid interface. Generally, the solution pH will remain constant because the anodic hydrolysis reaction produces excess H+ (or oxidizes OH− depending on solution pH). But to isolate the plasma effect, we also used the H-cell to separate the anodic and cathodic regions, such that local pH changes can be measured. This pH change can also be directly visualized using pH-sensitive dye [54], as shown in Fig. 5. Nominally, the dye is yellow under pH neutral conditions. But once the plasma cathode is activated, the solution turns green as it becomes more basic. Interestingly, the basic solution jets away from the interface, where it forms vortices upon impinging the bottom of the electrochemical cell. While this phenomenon is not completely understood, a preliminary analysis of the interfacial Debye layer that forms at the plasma–liquid interface has indicated that a strong electrokinetic flow is induced by the large flux of electrons injected into the solution by the plasma.

Copyright 2014 IEEE. Adapted, with permission, from [54]
Photograph of pH-sensitive dye used to visualize plasma-induced reduction reaction at the interface of a plasma cathode and liquid anode.
While these two studies confirmed that the plasma could directly reduce solution species and electrolyze water, it did not fully resolve whether other plasma-generated species are important. We explored this by studying the competition between plasma-induced electrolysis and other plasma–liquid effects using the model system of aqueous sodium chloride (NaCl) [55]. In a conventional electrochemical cell, electrolytic reactions in aqueous NaCl produce chlorine gas Cl2 in addition to H2 and O2. In particular, in addition to the water electrolysis reactions, a second reaction occurs at the anode via
The net result of this competition is an excess of (OH−)aq in the solution such that not just the local but the bulk pH increases. This is the classic chlor-alkali process used commercially to produce sodium hydroxide (NaOH). However, as noted earlier, the plasma generates a wide variety of reactive neutrally charged radicals. Thus, in addition to the electron transfer needed to maintain the DC plasma current, these neutral plasma species also dissolve into the solution. In particular, in nitrogen/oxygen-rich environments such as air, the plasma forms nitric oxides (NOx), and these plasma products dissolve into the solution to form nitrous (HNO2) and nitric (HNO3) acid by way of
Thus, the dissolved plasma species will decrease the pH of the bulk solution, in direct competition with the chlor-alkali process that increases the pH. Additionally, H2O2 is also produced which is weakly acidic. The NaCl system, therefore, is a nice platform to study how both electrolytic and dissolved plasma species compete during plasma–liquid interactions.
In order to resolve these effects, we varied the gas environment in the headspace above the solution where the plasma was formed [55]. In nitrogen-free environments, such as pure Ar and pure O2, the pH should increase as electrolytic processes dominate. In nitrogen-rich environments, such as pure N2 and air, the pH should decrease due to Reactions (4a) and (4b). Figure 6 shows the pH as a function of the plasma processing time along with pH measurements for a conventional electrochemical cell using a solution of 0.34 M NaCl. As expected, for the conventional electrochemical cell, there is only the chlor-alkali process and the pH increases. Further, the pH also increased for the pure Ar and pure O2 gas environments, although the increase was slightly smaller than that observed for the conventional cell. The reason is that even in the absence of N2, H2O2 is formed and is slightly acidic lowering the pH. The presence of H2O2 was subsequently confirmed using chemical assay analyses. In contrast, when the plasma was formed in pure N2 and air, the pH decreased significantly as HNO2 and HNO2 were generated, which was confirmed via ion chromatography. Curiously, the pH decrease was greater for pure air than for N2. The reason for this discrepancy is that NOx formation requires oxygen, which is not theoretically present in a pure N2 plasma. Here, the oxygen comes from the oxidation reaction at the anode that completes electrolysis. That is, in pure N2, the production of HNO2 and HNO3 is inherently limited by the electrolytic processes required to produce O2. Thus, while the electrolytic and dissolved plasma species can compete with each other, this showed that they also couple with each other to fundamentally alter the chemistry in the solution [55].

Reprinted with permission from [55]. Copyright 2013 American Chemical Society
Measured pH change for various plasma processing times when the plasma is formed in different gas environments (Ar, O2, N2, and air) as well as that for a conventional electrochemical cell.
3.3 Direct Detection of Solvated Electrons at the Plasma–Liquid Interface
While the aforementioned studies have provided insight to the nature of electron-driven reactions in plasma cathode configurations, they are all indirect—measuring long-lived products of electron-induced reactions. Thus, they provide little information about the complex reaction pathways and short-lived radicals that ultimately yield the stable bulk products. Recently, our group has developed an optical measurement to directly detect plasma-injected solvated electrons at the plasma–liquid interface [56].
Solvated electrons exhibit a broad optical absorption spectrum that peaks around 700 nm at room temperature [57]. They are typically produced and studied via radiolysis experiments, where high-energy radiation ionizes an aqueous solution throughout the bulk of a reactor vessel. The ample amount of solvated electrons produced by radiolysis makes optical measurements relatively simple, and the rate constants for hundreds of different reactions involving (e−)aq have been measured and tabulated [52].
In contrast to radiolysis, the solvated electrons in the plasma cathode electrochemical configurations are injected into the solution across an interface, rather than being produced volumetrically in the bulk solution. Due to Reactions (1) and (2), the solvated electrons only penetrate a few nanometers into the solution before reacting away. This makes for a very short optical path length and an optical absorbance on the order of 10 ppm. To measure the small optical signal, we utilized a total internal reflection geometry along with lock-in amplification as illustrated in Fig. 7a. With this total internal reflection absorption spectroscopy (TIRAS) technique, we were able to measure the absorption spectrum of the interfacial solvated electrons as shown in Fig. 7b and examine their reaction kinetics with known scavengers such as \({\text{NO}}_2^ - ,{\text{ NO}}_3^ - ,\) H2O2, and H+ (as shown in Table 1).

Used with permission from [56]
a Schematic of the total internal reflection absorption spectroscopy (TIRAS) configuration used to measure solvated electrons. b The optical absorption spectrum of solvated electrons measured at the plasma–liquid interface compared to the well-known bulk spectrum (solid black line).
Interestingly, both the measured optical absorption spectrum and the measured reaction rate coefficients are similar to those generated via pulse radiolysis. However, there are important differences, particularly in the case of the absorption spectrum. For the plasma–liquid interface, it is apparently shifted to higher energies (to the blue) and the Lorentzian blue tail appears to be suppressed. These differences are likely attributable to the unique environment that exists at the plasma–liquid interface in contrast to the environment in bulk radiolysis. In particular, there is an electric double layer that forms on both the plasma side and the liquid side, each of which boast significant space charge as well as an electric field. Radiolysis experiments, in contrast, are typically in electrically neutral solutions with no electric field. These effects could be responsible for the observed differences in absorption as well as reaction rate coefficients, but it is clear that more study is required.
As atmospheric pressure plasmas are useful because they operate in ambient air, we also looked at the effect of air on the total yield of solvated electrons [59]. These results showed that O2(g) in the plasma greatly lowers the concentration of solvated electrons. This is likely because O2(g) is electronegative and scavenges free electrons in the plasma phase to form \({\text{O}}_{2({\text{g}})}^ - ,\) and then the remaining electrons that do solvate are quickly scavenged by reactive oxygen species in the liquid phase. Therefore, in atmospheric air plasma electrochemistry configurations, solvated electrons themselves may play a minor role in reducing particular solution species, but appear to be an important intermediate in the various pathways of ROS.
4 Opportunities for Catalyst-Free Plasma Electrochemical Processing
These studies have revealed important fundamental aspects of the electrochemical behaviour of plasma cathode electrochemical systems—(1) the plasma injects electrons into the solution to initiate reactions, and (2) these electrons solvate and behave like solvated electrons generated by radiolysis. What these studies reveal is that the behaviour at the plasma cathode/liquid interface is more closely aligned with radiation chemistry than electrochemistry. As noted by Sankaran’s group, eliminating the cathode has potential technological advances, primarily eliminating the need for expensive or exotic electrocatalyst materials [51]. In essence, using a plasma cathode system creates the opportunity for catalyst-free electrochemical processing. However, the detection and measurement of plasma-solvated electrons [56] suggests that using plasma cathode systems for chemical processing should be guided more by radiation chemistry than electrochemistry.
We have recently taken this approach for the plasma electrochemical processing of carbon dioxide (CO2) [60], building on the suggestion of Sankaran’s group [51]. Electrochemical processing of CO2 is a promising way to convert what is often considered our most significant pollutant into other useful chemicals, including industrial chemicals like formic acid (HCOOH), clean-burning hydrocarbons such as methanol (CH3OH), and synthetic gas (CO + H2) for further downstream refinement [1]. Our preliminary results have shown that the plasma can electrochemically reduce (CO2)aq to formic acid and oxalate with a Faradaic efficiency of ~10%. This is relatively low when compared with recent electrocatalytic techniques, as shown in Table 2. However, given the known reaction kinetics of solvated electrons, we are highly optimistic that our method can be refined to reach Faradaic efficiencies above 90% with a very high selectivity.
In a plasma electrochemical system, the CO2 is dissolved into solution and the plasma is brought into contact as a plasma cathode. The first chemical step is anticipated to be direct reduction by a solvated electron to form the carboxyl radical anion \({\text{CO}}_2^{ \bullet - },\)
Using our TIRAS measurement (Fig. 7a), the concentration of solvated electrons monotonically decreases as the concentration of dissolved CO2 increases as shown in Fig. 8a. This non-linear behavior is consistent with the first order reaction kinetics suggested by Reaction (5) and from it, the reaction rate coefficient can be calculated to be k = 7.4 × 109 M−1 s−1 [60]. Again, this value compares well with the value of 7.7 × 109 M−1 s−1 reported in the radiation chemistry literature [64], indicating that the plasma electrochemical system behaves like a radiolysis system even though the plasma itself does not ionize the solution.

Used with permission from [60]
a The measured absorbance as a function of increasing dissolved CO2 concentration. Ion chromatographs of the products of plasma processing 34 mM of dissolved CO2 for approximately 5 min at 10 mA in solutions of b 20 mM sodium hydroxide (NaOH) and c 2 nM of sulfuric acid (H2SO4).
Since we anticipated the solution to behave like a radiolysis system, we utilized radiolysis literature to highlight the subsequent reaction pathways of the carboxyl radical anion. At high pH, we anticipated recombination to form oxalate via [65, 66]
whereas at low pH we anticipated the equilibrium reaction
leading to disproportionation and ultimately formate (HCOO−) [67]. By varying the pH of our solution using either sodium hydroxide (NaOH) or sulfuric acid (H2SO4), we could measure the products after plasma processing. As shown in Fig. 8b, c, we detect oxalate under basic conditions and both oxalate and formate under acidic conditions. This was not only consistent with our expectations, but also highlights that some level of selectivity is possible using catalyst-free plasma electrochemistry if the solution chemistry is properly controlled.
As Reaction (5) is the first step in either pathway that forms oxalate or formate, we can turn to the radiation chemistry kinetics to predict the reduction efficiency. With a rate of \({k_{{\text{C}}{{\text{O}}_{\text{2}}}}}={\text{7}}.{\text{4}} \times {\text{1}}{0^{\text{9}}}{\text{ }}{{\text{M}}^{ - {\text{1}}}}{\text{ }}{{\text{s}}^{ - {\text{1}}}}\) and a dissolved CO2 concentration of [(CO2)aq] = 34 mM, we can predict that CO2 reduction—Reaction (5)—will dominate second order recombination—Reaction (2)—and we should have >90% faradaic efficiency for \({\text{CO}}_2^{ \bullet - }.\) (That is, \({k_{{\text{C}}{{\text{O}}_2}}}{\left[ {{{\left( {{{\text{e}}^ - }} \right)}_{{\text{aq}}}}\left] {\left[ {{{\left( {{\text{C}}{{\text{O}}_{\text{2}}}} \right)}_{{\text{aq}}}}} \right] \gg {\text{2}}k_{e}} \right[{{\left( {{{\text{e}}^ - }} \right)}_{{\text{aq}}}}} \right]_{\text{2}}}\)). However, our measurements of oxalate and formate yield indicated only ~8–9% faradaic efficiency; much less than the 90% predicted by the kinetics. There are two possible reasons for this. One is that there could be other solvated electron pathways that compete with Reaction (5) that limit CO2 reduction. As the plasma undoubtedly creates other dissolved species, including OH· and H2O2, that can react with solvated electrons, this remains a possibility. But these species would have to be on the order of the 34 mM of dissolved CO2 to truly compete for solvated electrons. The other possibility is that there are alternative chemical pathways for the carboxyl radical anion \({\text{CO}}_2^{ \bullet - }\) as well as for \({({\text{CO}}_2^ - )_2}\) and CO2H· that went undetected and would affect the yield of formate and oxalate. Future work should focus on uncovering other potential products that are initiated by the carboxyl radical anion. Regardless, this work demonstrated that using radiation chemistry to inform plasma electrochemistry, catalyst-free electrochemical processing is possible.
5 Discussion and Conclusions
Electrocatalysis is an exciting field that offers a significant number of opportunities for chemical processing. However, using plasma electrochemical systems offers an alternative approach where the chemistry is dictated not by the catalytic performance of the electrodes but the free radical chemistry of plasma-injected species. With the ubiquitousness of atmospheric pressure plasma systems, this approach to electrochemistry is not only cost-effective, but relatively simple to implement. In this paper, we have overviewed recent efforts to both utilize and understand these systems, highlighting their potential promise for materials synthesis and CO2 reforming. Importantly, the plasma–liquid interface is a complex environment, and its chemistry is dictated by many short-lived radical species. Knowing the precise chemical pathways and intermediates that yield various products alleviates much of the trial-and-error in designing systems targeted to various applications. For example, now that it is understood that solvated electrons are the primary reducing agent in a liquid anode (plasma cathode) configuration, designing a chemical synthesis procedure simply becomes a matter of looking through the radiolysis literature [52] for the desired reaction and its kinetics.
Moving forward, it is necessary to develop more sophisticated measurement techniques that directly probe the interface for various short-lived intermediate species as well as other interfacial properties. As shown here, it is possible to perform very precise measurements of the interface itself using lock-in amplification. Many plasma sources are already AC-driven, making it easier to apply sensitive lock-in amplification techniques. While our own work has primarily focused on solvated electrons [56, 59], there are many other free radicals such as (\({\text{O}}_2^ -\))aq, (HO2)aq, and (O3)aq, that are critical in many applications and can potentially be measured by their optical absorption spectrum [68].
In addition to fundamental studies on the plasma–liquid interface, new applications can and should be developed. While materials synthesis is very promising, much work is still required to control the size distribution and morphology of synthesized nanoparticles, as well as to use the technique for non-metal materials such as semiconductors. For other types of electrolytic processing, our work has shown CO2 as a promising approach but other applications—whether they rely on plasma-induced reduction or oxidation—still require exploration.
References
Qiao J, Liu Y, Hong F, Zhang J (2014) A review of catalysts for the electro reduction of carbon dioxide to produce low-carbon fuels. Chem Soc Rev 43:631
Ni M, MKH Leung, DYC Leung, Sumathy K (2007) A review and recent developments in photocatalytic water-splitting using TiO2 for hydrogen production. Renew Sustain Energy Rev 11:401
Liu H, Song C, Zhang L, Zhang J, Wang H, Wilkinson DP (2006) A review of anode catalysis in the direct methanol fuel cell. J Power Sources 155:95
SKS Gupta (2015) Contact glow discharge electrolysis: its origin, plasma diagnostics and non-faradaic chemical effects. Plasma Sources Sci Technol 24:063001
Cavendish H (1784) Experiments on air. Phil Trans Royal Soc London 75:372
Rayleigh L, Ramsay W (1895) Argon, a new constituent of the atmosphere. Proc Roy Soc London 57:265
Schönbein CF (1840) Beobachtungen über den bei der Elektrolysation des Wassers und dem Ausströmen der gewöhnliehen Elektricität aus Spitzen sich entwikkelnden Geruch. Ann Phys 126:616
Birkeland KR (1889) On the oxidation of atmospheric nitrogen in electric arcs. Nature 58:98
Gubkin J (1887) Electrolytische Metallabscheidung an der freien Oberfläche einer Salzlösung. Ann Phys Chem 32:114
Klemenc A (1927) Zur Kenntnis der elektrolytischen Reduktion und der Reaktionen im Glimmbogen an der Phasengrenze Flüssigkeit-Gas. Z Physik Chem A 130:378
Bruggeman P, Leys C (2009) Non-thermal plasmas in and in contact with liquids. J Phys D 42:053001
Mariotti D, Sankaran RM (2010) Microplasmas for nanomaterials synthesis. J Phys D 43:323001
Hickling A (1971) Electrochemical processes in glow discharge at the gas-solution interface. In: Bockris JO’M, Conway BE (eds) Modern aspects of electrochemistry, vol 6. Springer, New York, pp 329–373
Denaro AR, Hickling A (1958) Glow-discharge electrolysis in aqueous solutions. J Electrochem Soc 105:265
Cserfalvi T, Mezei P, Apai P (1993) Emission studies on a glow discharge in atmospheric pressure air using water as a cathode. J Phys D 26:2184
Mezei P, Cserfalvi T (2007) Electrolyte cathode atmospheric glow discharges for direct solution analysis. Appl Spectrosc Rev 42:573
Webb MR, Hieftje GM (2009) Spectrochemical analysis by using discharge devices with solution electrodes. Anal Chem 81:862
Jamorz P, Greda K, Pohl P (2012) Development of direct-current, atmospheric-pressure, glow discharges generated in contact with flowing electrolyte solutions for elemental analysis by optical emission spectrometry. Trends Anal Chem 41:105
Schwarz AJ, Ray SJ, Hieftje GM (2015) Automatable on-line generation of calibration curves and standard additions in solution-cathode glow discharge optical emission spectrometry. Spectrochim Acta Part B 105:77–83
Richmonds C, Sankaran RM (2008) Plasma-solution electrochemistry: rapid synthesis of colloidal metal NPs by microplasma reduction of aqueous cations. Appl Phys Lett 93:131501
Chen Q, Kaneko T, Hatakeyama R (2012) Reductants in gold nanoparticle synthesis using gas–liquid interfacial discharge plasmas. Appl Phys Express 5:086201
Patel J, Nemcova L, Maguire P, Graham WG, Mariotti D (2013) Synthesis of surfactant-free electrostatically stabilized gold nanoparticles by plasma-inducted liquid chemistry. Nanotechnology 24:245604
Shirai N, Uchida S, Tochikubo F (2014) Synthesis of metal nanoparticles by dual plasma electrolysis using atmospheric dc glow discharge in contact with liquid. Jpn J Appl Phys 53:046202
Brettholle M, Hofft O, Klarhofer L, Mathes S, Maus-Friedrichs W, Zein El Abedin S, Krischok S, Janek J, Endres F (2010) Plasma electrochemistry in ionic liquids: deposition of copper nanoparticles. Phys Chem Chem Phys 12:1750–1755
Kulbe N, Hofft O, Ulbrich A, Zein El Abedin S, Krischok S, Janek J, Polleth M, Endres F (2011) Plasma electrochemistry in 1-butyl-3-methylimidazolium dicyanamide: copper nanoparticle from CuCl and CuCl2. Plasma Processes Polym 8:32–37
Hofft O, Endres F (2011) Plasma electrochemistry in ionic liquids: an alternative route to generate nanoparticles. Phys Chem Chem Phys 13:13472–13478
Kaneko T, Baba K, Hatakeyama R (2009) Gas–liquid interfacial plasmas: basic properties and applications to nanomaterial synthesis. Plasma Phys Control Fusion 51:124011
Lee SW, Janyasupab M, Liu C-C, Sankaran RM (2013) Fabrication of Ir nanoparticle-based biosensors by plasma electrochemical reduction for enzyme-free detection of hydrogen peroxide. Catal Today 211:137–142
Endres F, Hofft O, von Brisinski NS (2014) Plasma electrochemistry in ionic liquids: from silver to silicon nanoparticles. J Mol Liq 192:59–66
Ghosh S, Bishop B, Morrison I, Akolkar R, Scherson D, Sankaran RM (2015) Generation of a direct-current, atmospheric-pressure microplasma at the surface of a liquid water microjet for continuous plasma–liquid processing. J Vaccum Sci Technol A 33:021312
Mariotti D, Švrček V, Hamilton JW, Schmidt M, Kondo M (2012) Silicon nanocrystals in liquid media: optical properties and surface stabilization by microplasma-induced non-equilibrium liquid chemistry. Adv Funct Mater 22:954–964
McKenna J, Patel J, Mitra S, Soin N, Švrček V, Maguire P, Mariotti D (2011) Synthesis and surface engineering of nanomaterials by atmospheric-pressure microplasmas. Euro Phys J 56:24020
Chen Q, Li J, Li Y (2015) A review of plasma-liquid interactions for nanomaterial synthesis. J Phys D 48:424005
Mezei P, Cserfalvi T (2012) A critical review of published data on the gas temperature and the electron density in the electrolyte cathode atmospheric glow discharges. Sensors 12:6576–6586
Oehmigen K, Hähnel M, Brandenburg R, Wilke C, Weltmann K-D, von Woedtke T (2010) The role of acidification for antimicrobial activity of atmospheric pressure plasma in liquids. Plasma Process Polym 7:250
CAJ van Gils, Hofmann S, BKHL Boekema, Brandenburg R, Bruggeman PJ (2013) Mechanisms of bacterial inactivation in the liquid phase induced by a remote RF cold atmospheric pressure plasma jet. J Phys D 46:175203
Locke BR, Shih K-Y (2011) Review of the methods to form hydrogen peroxide in electrical discharge plasma with liquid water. Plasma Sources Sci Technol 20:034006
Vasko CA, Liu DX, vanVeldhuizen EM, Iza F, Bruggeman PJ (2014) Hydrogen peroxide production in an atmospheric pressure RF glow discharge: comparison of models and experiments. Plasma Chem Plasma Process 34:1081–1099
Graves DB (2012) The emerging role of reactive oxygen and nitrogen species in redox biology and some implications for plasma applications to medicine and biology. J Phys D 45:263001
Norberg SA, Tian W, Johnsen E, Kushner MJ (2014) Atmospheric pressure plasma jets interacting with liquid covered tissue: touching and not-touching the liquid. J Phys D 47:475203
Liu ZC, Liu DX, Chen C, Li D, Yang AJ, Rong MZ, Chen HL, Kong MG (2015) Physicochemical processes in the indirect interaction between surface air plasma and deionized water. J Phys D 48:495201
Liu DX, Liu ZC, Chen C, Yang AJ, Li D, Rong MZ, Chen HL, Shama G, Kong MG (2016) Aqueous reactive species induced by a surface air discharge: heterogeneous mass transfer and liquid chemistry pathways. Sci Rep 6:23737
Lindsey AD, Graves DB, Shannon SC (2016) Fully coupled simulation of the plasma liquid interface and interfacial coefficient effects. J Phys D 49:235204
Yusupov M, Neyts EC, Simon P, Berdiyorov G, Snoeckx R, ACT van Duin, Bogaerts A (2014) Reactive molecular dynamics simulations of oxygen species in a liquid water layer of interest for plasma medicine. J Phys D 47:025205
T Cserfalvi, P Mezei (1996) Operating mechanism of the electrolyte cathode atmospheric glow discharge. Fresen J Anal Chem 355:813–819
Denaro AR, Hough KO (1972) Glow-discharge electrolysis of sulphuric acid solutions. Electrochim Acta 17:549
Khlyustova AV, Maksimov AL, Khorev MS (2008) Radiation of metal atoms in the plasma of an atmospheric pressure glow discharge with an electrolyte cathode. Surf Eng Appl Electrochem 44:370
Marcus RK, Davis WC (2001) An atmospheric pressure glow discharge optical emission source for the direct sampling of liquid media. Anal Chem 73:2903–2910
Schwartz AJ, Ray SJ, Elish E, Storey AP, Rubinshtein AA, Chan GC, Pfeuffer KP, Hieftje GM (2012) Visual observations of an atmospheric-pressure solution-cathode glow discharge. Talanta 102:26
Quarles CD, Gonzalez J, Choi I, Ruiz J, Mao X, Marcus RK, Russo RE (2012) Liquid sampling-atmospheric pressure glow discharge optical emission spectroscopy detection of laser ablation produced particles: a feasibility study. Spectrochim Acta Part B 76:190
Richmonds C, Witzke M, Bartling B, Lee SW, Wainright J, Liu C-C, Sankaran RM (2011) Electron-transfer reactions at the plasma–liquid interface. J Am Chem Soc 133:17582
Buxton GV, Greenstock CL, Helman WP, Ross AB (1988) Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/O–) in aqueous solution. J Phys Chem Ref Data 17:513–886
Witzke M, Rumbach P, Go DB, Sankaran RM (2012) Evidence for the electrolysis of water by plasmas formed at the surface of aqueous solutions. J Phys D 45:44201
Rumbach P, Griggs N, Sankaran RM, Go DB (2014) Visualization of electrolytic reactions at a plasma–liquid interface. IEEE Trans Plasma Sci 42:2610
Rumbach P, Witzke M, Sankaran RM, Go DB (2013) Decoupling interfacial reactions between plasmas and liquids: charge transfer vs. plasma neutral reactions. J Am Chem Soc 135:16264
Rumbach P, Bartels DM, Sankaran RM, Go DB (2015) The solvation of electrons by an atmospheric pressure plasma. Nat Commun 6:7248
Bartels DM, Takahashi K, Cline JA, Marin TW, Jonah CD (2005) Pulse radiolysis of supercritical water. 3. Spectrum and thermodynamics of the hydrated electron. J Phys Chem A 109:1299
Rumbach P, Bartels DM, Sankaran RM, Go DB, Corrigendum (2015) The solvation of electrons by an atmospheric-pressure plasma. Nat Commun 7:11911
Rumbach P, Bartels DM, Sankaran RM, Go DB (2015) The effect of air on solvated electron chemistry at a plasma/liquid interface. J Phys D 48:424100
Rumbach P, Xu R, Go DB (2016) Electrochemical production of oxalate and formate from CO2 by solvated electrons produced using an atmospheric-pressure plasma. J Electrochem Soc 163:F1157–F1161
Lu Q, Rosen J, Zhou Y, Hutchings GS, Kimmel YC, Chen JG, Jiao F (2014) A selective and efficient electrocatalyst for carbon dioxide reduction. Nat Commun 5
Angamuthu R, Byers P, Lutz M, Spek AL, Bouwman E (2010) Electrocatalytic CO2 conversion to oxalate by a copper complex. Science 327:313–315
Nakata K, Ozaki T, Terashima C, Fujishima A, Einaga Y (2014) High-yield electrochemical production of formaldehyde from CO2 and seawater. Angew Chem 126:890–893
Gordon S, Hart EJ, Matheson MS, Rabani J, Thomas JK (1963) Reactions of the hydrated electron. Discuss Faraday Soc 36:193–205
Fojtik A, Czapski G, Henglein A (1970) Pulse radiolytic investigation of the carboxyl radical in aqueous solution. J Phys Chem 74:3204–3208
Lin M, Katsumura Y, Muroya Y, He H, Miyazaki T, Hiroishi D (2008) Pulse radiolysis of sodium formate aqueous solution up to 400 °C: absorption spectra, kinetics and yield of carboxyl radical \({\text{CO}}_2^{ \bullet - }\). Rad Phys Chem 77:1208–1212
Flyunt R, Schuchmann MN, von Sonntag C (2001) A common carbanion intermediate in the recombination and proton-catalysed disproportionation of the carboxyl radical anion, \({\text{CO}}_2^{ \bullet - },\) in aqueous solution. Chem Eur J 7:796–799
Bhüler RE, Staehelin J, Hoigné J (1984) Ozone decomposition in water studied by pulse radiolysis. 1. Perhydroxyl (HO2)/hyperoxide (\({\text{O}}_2^ -\)) and \({\text{H}}{{\text{O}}_{\text{3}}}/{\text{O}}_3^ -\) as intermediates. J Phys Chem 88:2560–2564
Acknowledgements
We would like to acknowledge our collaborators R. Mohan Sankaran, David M. Bartels, and Megan Witzke who have contributed to our work this area. R. Mohan Sankaran also provided Fig. 2 in this manuscript. The authors’ work has been supported by the US Army Research Office under Award Number W911NF-14-1-0241 and the Electrochemical Society Toyota Young Investigator Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rumbach, P., Go, D.B. Perspectives on Plasmas in Contact with Liquids for Chemical Processing and Materials Synthesis. Top Catal 60, 799–811 (2017). https://doi.org/10.1007/s11244-017-0745-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-017-0745-9