
Abstract
Interactions between plasmas and liquids lead to the formation of a variety of unique reactive species for chemical and materials applications that makes plasma-induced liquid chemistry attractive for industrial applications. While formation of various chemical species have been observed, a complete understanding of the chemistry occurring at the plasma–liquid interface remains unclear. Here, we study the properties of water exposed to a helium plasma at atmospheric pressure in open air and in argon-controlled environments with systematic changes to process conditions. The pH, temperature, and conductivity are monitored as a function of plasma current and processing time. In addition, molecular oxygen (O2) and hydrogen peroxide (H2O2) are quantitatively measured. We find that the electron density which is controlled by plasma current plays an important role and the number of electrons injected from the plasma into water can be related to the number of H2O2 molecules generated. We support this result with a mechanistic description of reaction pathways at the plasma-water interface.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Plasmas formed inside liquids and at the surface of liquids have become of increasing interest in the last few years, mainly due to their practical applications in many areas [1–3]. A number of investigations have addressed reaction mechanisms taking place in plasma-liquid systems and species present both in the gas and in the liquid phase have been characterized [4]. Chemical reactions occurring near the plasma–liquid interface are of particular importance because of unique reactive species that could be important in biological [5], chemical [6], and materials [7] applications. However, the interfacial reactions are highly complex and system-dependent because of the different gas environments, electrode configurations, and solutions that have been studied, making it difficult to obtain clear, in-depth understanding of the reaction mechanisms [8]. In general, plasma–liquid interactions generate a variety of different energetic species (depending on the type of plasmas and liquids) such as hydrogen peroxide (H2O2), hydroxyl radicals (OH•), hydroperoxyl radicals (HO2), hydrogen radicals (H•), oxygen radicals (O•), ozone (O3) [2, 4, 9]. Different techniques have been used to analyze the generated species either in the gas or liquid phase, including pH and conductivity measurements [10], optical emission spectroscopy (OES) [11], gas chromatography [12], mass spectrometry [13], cyclic voltammetry [14], and UV–Vis absorbance spectroscopy [15]. In particular, pH and conductivity measurements are simple and easy to implement, but can provide important insight. Identifying the presence and nature of various chemical species in the liquid can be a leading key to understand the plasma-activated chemistry. In the present study, we characterized changes in pH, temperature, and conductivity of water (distilled water with an initial conductivity of 1.9 μS cm–1) exposed to a plasma as a function of time, and correlated the changes to the formation of oxygen molecules and hydrogen peroxide (H2O2) which were directly measured by spectroscopic techniques. Results were compared for a background of air versus argon (Ar) in order to understand the effect of oxygen (O2) and nitrogen (N2) in air on reaction chemistry. We find that the acidity of water increases with current and time of plasma processing and the atmospheric air surrounding the plasma affects the pH level of water during treatment. We also show that H2O2 concentrations are found to be current-dependent and the backgrounds of air and Ar indeed affect the H2O2 concentrations in water. From these measurements, we are able to hypothesize possible reaction channels at the plasma-liquid interface. While the study is again system-specific, the plasma-aqueous solution interface that is studied here is one of the most widely encountered chemistries and, therefore, has wide applicability.
EXPERIMENTAL
The schematic of the setup used for the plasma-water experiments is shown in Fig. 1. The electrodes consisted of a grounded stainless steel capillary tube with an outer diameter of 0.5 mm and an internal diameter of 0.25 mm held at ~0.7 mm above the water surface serving as the cathode, and a 6 mm diameter carbon rod immersed in the water bath serving as the anode. The capillary tube was pressurized with helium (He) gas to a flow rate of 25 standard cubic centimeters (sccm). The plasma was powered by a direct current power supply (Glassman High Voltage, Inc.). When a positive voltage of >2.8 kV was applied, a microplasma was ignited in the He gas flow between the end of the capillary tube and the water surface. The applied voltage was varied to maintain the current constant in experiments.

Schematic of the experimental setup used to study plasma–water interactions; DC is direct-current.
Distilled water from a water distillation facility with a pH of ~5 and a conductivity of 1.9 μS cm–1 was used. No additional ions were added. The small acidity and conductivity suggest that the water contained dissolved carbon dioxide (CO2) which forms carbonic acid (H2O + CO2 \( \leftrightarrow \) H2CO3 \( \leftrightarrow \) H+ + \({\text{HCO}}_{3}^{ - }\)) [16]. The distilled water (20 mL) was placed in a 5 cm diameter glass Petri dish and exposed to the atmospheric-pressure microplasma at currents of 2.5 and 5 mA, respectively, for 2, 6, and 10 min. The corresponding applied voltages were varied between 2.8 and 0.9 kV to produce a constant current of 2.5 mA, and between 3.5 and 1.1 kV to produce a constant current of 5 mA; as discussed later, it was necessary to decrease the applied voltage to maintain a constant current because of an increase in the solution conductivity. Additional details of the electrical characteristics and equivalent circuit model are detailed in the Supplementary Information. After treating the water, the solution pH, temperature, and conductivity were measured. In addition, O2 in the gas phase, dissolved O2, and H2O2 were detected. The temperature was measured using an HI 7662 stainless steel temperature probe with an NTC sensor. The pH and conductivity were measured with a 212 pH meter from Hanna Instruments, 712 conductometer from Metrohm, and an EC500 pH/conductivity meter from Extech Instruments Co. The temperature was an average value obtained by immediately placing a probe in the approximate area where the plasma interfaced the liquid after the plasma was turned off. The position of the probe was kept constant to provide a reference location and the measurement was not intended to produce a spatially resolved description of the water temperature. The H2O2 concentration was measured spectroscopically by reaction with titanyl ions which yields pertitanic acid, a yellow-colored complex with a distinct absorption band at ~407 nm [17, 18]. The reaction was carried out by adding a solution of titanium(IV) oxysulfate in sulfuric acid (Ti–S) (Sigma Aldrich, UK) to the plasma-treated water in a volume ratio of 1 : 2. Absorbance spectra were acquired by a Lambda 35 UV–Vis spectrometer from Perkin Elmer. Because absorbance is measured quantitatively, the H2O2 concentration could be determined from the intensity of the absorbance at 407 nm [18]. Calibration curves were constructed to relate the measured values to the H2O2 concentration. H2O2 measurements were all carried out shortly after processing; nonetheless, we have verified that the H2O2 concentration remained stable after processing for several hours. The relative O2 concentration in the gas phase was measured near the plasma–water interface by using a Unisense microsensor multimeter (detection limit of 0.3 µM). The device relates the O2 concentration to an electrical signal measured (in mV). A dissolved oxygen meter (DO 600, Extech Instruments Co.) was used to measure the dissolved O2 concentration in the water after each treatment.
RESULTS AND DISCUSSIONS
Temperature Measurements
Temperature measurements showed that the water temperature increased with increasing plasma processing time (Fig. 2). The temperature initially increased rapidly. In the first 2 min, the temperature was found to increase by 5 and 14°C for 2.5 and 5 mA current, respectively. After 2 min, the temperature continued to increase, but not as fast, at a similar rate of ~0.6 K/min for both currents. Although the temperature increased, the evaporation losses were minimal, ~0.05% (by volume) per minute.

Temperature of water exposed to plasma at constant currents of 2.5 and 5 mA as a function of time.
The observed increase in the temperature of the water after exposure to the plasma is not surprising. The neutral gas temperature of atmospheric-pressure microplasmas has been estimated to be on the order of hundreds of Kelvins and as high as 1000 K [19]. However, despite the high temperature, the plasma-water interface is relatively small compared to the volume of the water bath and heat from the microplasma is easily dissipated. Another possible mechanism for the heating of the bath is ohmic heating. To assess the role of ohmic heating, we carried out experiments with a standard electrochemical setup, i.e., the stainless steel capillary was in physical contact with the liquid to allow current flow without forming a plasma. We found that the water temperature increased with a very similar trend (an increase of 18°C at a current of 5 mA after 10 min) demonstrating that ohmic heating is the main factor contributing to the temperature increase. Nonetheless, we note that strong temperature gradients are expected in the proximity of the plasma–water interface which are very likely balanced by convective cooling both in the gas and liquid phase, leading to the lower average temperature.
pH and Conductivity Measurements
The pH was found to decrease after plasma exposure, changing more rapidly in the first 2 min and with a much more pronounced slope at 5 mA as compared to 2.5 mA current (Fig. 3).

Variation in pH and conductivity of water exposed to plasma at 2.5 and 5 mA as a function of time.
The conductivity was found to follow the pH (see Fig. 3). This aspect will be further discussed below. A common feature of the temperature, pH, and conductivity changes is a strong dependence on discharge current.
Oxygen Measurements
The relative O2 concentration in the gas phase above the water surface was found to increase with time (Fig. 4a). The oxygen concentration returned quickly to the initial value once the plasma was turned off. The dissolved oxygen concentration in water also increased over time (Fig. 4b). Increasing the current from 2.5 to 5 mA enhanced the oxygen production rate in water.

(a) Time dependence of the relative molecular oxygen concentration in the gas phase measured just above the plasma–water interface. The plasma was operated at 5 mA current for 5 min and then shut off. (b) Time dependence of dissolved oxygen concentration in plasma treated water at a current of 2.5 and 5 mA.
The formation of O2 gas as result of plasma–water interactions has been previously reported [20–25]. Multiple reaction paths are possible for the generation of O2. One type of reaction may involve charged and excited species in the plasma dissociating water vapor to produce gas phase radicals. Previous experiments have shown singlet delta oxygen O2 (\(^{1}{{\Delta }_{g}}\)) and superoxide \({\text{O}}_{2}^{{ - \bullet }}\) in plasmas containing water vapor [21, 26]. Alternatively, electrons from the plasma that interact with water molecules either at the gas side of the plasma–liquid interface, or at the liquid side as hydrated electrons by solvating, may react with water molecules to form a range of radicals (e.g., H, OH, O) leading to molecular oxygen as result of cascaded chemistry within the bulk of the liquid. Solvated electrons are known to be one of the most reactive species in all of chemistry [27]. Another possibility is that O2 gas is generated at the metal anode by water electrolysis. As we show in our analysis below, the main contribution to O2 formation is believed to be water oxidation at the anode.
Hydrogen Peroxide Measurements
As previously described, H2O2 was spectroscopically detected and the concentration (in mM) was determined from calibration curves. It should be noted here that it is well known that H2O2 is not formed in a standard electrochemical cell which we also verified with our experimental setup; therefore, the formation of H2O2 assumes particular importance because it suggests that very different mechanisms are at play in plasma-induced liquid chemistry.
The H2O2 concentration was found to increase with time at an almost constant rate at process times larger than 2 min, with a higher rate at higher current (Fig. 5).

H2O2 concentration plotted as a function of processing time at two different plasma currents.
The observed increase in H2O2 concentration over time is consistent with previously reported results [10, 28, 29], although the system configurations were different. We extended these experiments to other currents and found that the H2O2 production rate showed a linear dependence on current above 0.10 mA (Fig. 6).

H2O2 concentration (mM) in plasma treated water as a function of plasma current. The process time was 10 min in all cases. Inset shows the H2O2 concentration at the lower currents.
The dependence of H2O2 production on the plasma current has been previously reported [25, 30–32]. The linear relationship suggests a first order process with electrons playing a key role since the electron density is linearly dependent on the current. We compared the number of H2O2 molecules produced to the number of electrons injected from the plasma, the latter of which is obtained by assuming that the current is directly related to the electron flux. This analysis reveals that on average, one H2O2 molecule is produced for every ten electrons injected into the water. The rate of H2O2 formation was greatest in the first 2 min with rates of ~72 µM min–1 at 5 mA, decreasing to a constant value of ~22 µM min–1, independent of the current, at longer times (see Fig. 5). The inset of Fig. 6 shows that at low plasma currents, the behavior is non-linear and hence may indicate that some threshold plasma conditions are required to activate the reactions which lead to H2O2 formation.
Experiments in Argon-Controlled Environment
The pH change and H2O2 produced in plasma treated water were measured in an argon-controlled environment and compared with the previous results for open air (Fig. 7).

Comparison of (a) pH and (b) hydrogen peroxide concentrations for the plasma treated water samples in argon-controlled environment and in open air. The water samples were treated at current of 2.5 mA for the time period up to 10 min.
The pH of the plasma treated water in argon-controlled experiment was found to be relatively constant (~5) as compared to the open air experiments where the pH gradually decreased over time (Fig. 7a). This indicates that the mechanism for acidification of the water involves air (i.e., O2 and/or N2) which we discuss in further detail later. The H2O2 concentration increased over time, eventually saturating at ~5 min. This confirms that O2 in air is not responsible for the formation of H2O2. In fact, the H2O2 concentration was two to three times higher in an argon-controlled environment than in open air. The ratio of the number of electrons injected to the number of H2O2 molecules produced was estimated to be ~2 in the first few minutes and increased to 5 at 10 min for the argon-controlled environment.
Mechanistic Analysis of Plasma-Induced Liquid Chemistry
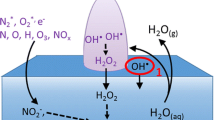
Based on the experimental results, we now provide a qualitative description of the reactions at the plasma–water interface that lead to the observed pH, temperature, and conductivity changes, as well as the O2 and H2O2 products (see Scheme 1). We note that the plasma–water interface is a highly complex and non-uniform environment, made up of a flowing He microplasma impinging on the water surface, an equilibrium layer of water vapor, and liquid water with ionic species (e.g., H+, OH–, \({\text{HCO}}_{3}^{ - }\)). For simplicity, we can consider that at some distance above the interface, a pure He plasma (no water vapor) exists. Similarly, at some distance below the interface, bulk liquid water exists. The He plasma also interacts with the surrounding air which diffuses in at the boundaries and can affect reactions occurring at the plasma–liquid interface. We discuss this issue later. Therefore, as a starting point, Scheme 1 is relevant to an argon-controlled environment where the contribution of air is ignored.

Schematic of possible reactions at plasma–water interface. For simplicity, we have omitted reducing reactions (e.g., H2 evolution), as these do not contribute to H2O2 formation.
Beginning at the anode (the immersed carbon rod), the corresponding charge transfer reactions should be identical to a standard electrochemical cell which lead to molecular O2 (at the liquid– solid electrode interface) via the following:
so that electrical continuity is maintained and charge neutrality in solution is preserved. This assumption is partially confirmed by the O2 measurements (Fig. 4). O2 in the gas phase could also originate from the liquid phase by the reaction
However this reaction is more likely to occur in a basic solution while in an acidic solution, as in our case (see Fig. 3), reaction (1) is more important.
At the cathode (plasma–liquid interface), several reactions are possible which we classify into three categories [3]: (i) reactions that occur in the gas phase resulting in radical species that subsequently react and/or become absorbed in the liquid; (ii) reactions that occur at the plasma-water interface involving plasma electrons (\({\text{e}}_{{{\text{gas}}}}^{ - }\)) still possessing some kinetic energy (e.g., dissociative electron attachment reactions); (iii) reactions within the liquid phase where electrons are essentially hydrated (or in other transient forms) [33, 34] and can be highly reactive.
Reactions of type (i) are different than those typically encountered in plasma chemical processes because electrons react with vapor supplied from (i.e., in equilibrium with) the liquid. The species produced via reactions (i) (i.e., \({{{\text{H}}}^{ \bullet }}\), \({\text{O}}{{{\text{H}}}^{ \bullet }}\), etc.) may react and/or be absorbed in the liquid. Direct ionization of water molecules is unlikely because of the high energy required (above ~12.6 eV) compared to direct electron dissociation (~5.1 eV) [21]. Since the O–H bond energy is ~5 eV, water molecules will split into different chemical species such as \({{{\text{H}}}^{ \bullet }}\), \({\text{O}}{{{\text{H}}}^{ \bullet }}\), \({{{\text{O}}}^{ \bullet }}\), etc. For instance \({{{\text{H}}}^{ \bullet }}\) and \({\text{O}}{{{\text{H}}}^{ \bullet }}\) radicals are formed via the following reaction [4]:
Reactions of type (ii) occur closer to the interface where electrons may be affected by the intermolecular potential of liquids. Dissociative electron attachment at the interface is an interesting type of reactions that could, for instance, lead to the following reaction (at plasma–liquid interface):
Here, subscript g/l indicates the species existing at gas–liquid interface. Electrons at the plasma–liquid interface may also become solvated (\({\text{e}}_{{{\text{aq}}}}^{ - }\)) when they reach the bulk liquid without inducing any reaction of type (ii), leading to reactions of type (iii). Although the formation of hydrated electrons remains a debated topic [35, 36], once formed, they are known to react very quickly with the most likely reaction (in liquid) being:
Direct reduction of H+ with hydrated electrons (2H+ + 2e– → H2 (all in liquid)) is also possible. However, direct reduction of hydrogen ions, even in a standard electrochemical cell, does not occur with hydrated electrons. In an electrochemical cell, reduction of hydrogen ions takes place either at the interface of the metal electrode (i.e., \(2{\text{H}}_{{{\text{(l)}}}}^{ + } + {\text{ }}2{\text{e}}_{{{\text{(s)}}}}^{ - }\) → H2(l)) or via OH– ions. As the metal–liquid interface is missing in the plasma–liquid cell, only hydrated electrons are available (i.e., \({\text{e}}_{{{\text{aq}}}}^{ - }\)) which are more likely to immediately react with water molecules (i.e., reaction (4)) before they can encounter hydrogen ions since the life time of hydrated electrons is very short and their penetration depth in water is limited [35–37].
Cascaded chemistry, defined here as coupled and multistep reactions involving different species and phases, will result from the proposed reactions induced at the plasma–liquid interface. In this study, we did not measure the production of molecular H2; however Witzke et al. [38] using a similar set-up have demonstrated that H2 gas is indeed produced from similar plasma-liquid systems. Therefore, it is reasonable to assume that hydrogen radicals produced by reaction (4) or dissolved in water from reaction (2) can form gas phase H2 by the following reaction (in liquid):
Negative hydrogen ions generated by reaction (3) will probably have very short lifetimes and rapidly react with water via the reaction H– + H2O → OH– + H2, reproducing species also formed by reactions (4) and (5). Overall, increased production of H+ and OH– ions contribute to the pH and conductivity changes.
The formation of H2O2 is a clear example of cascaded chemistry and can involve several different reaction paths. H2O2 may be formed via recombination of \({\text{O}}{{{\text{H}}}^{ \bullet }}\) radicals in the gas-phase and subsequent absorption of H2O2 in the liquid, which has been previously reported [39, 40]. H2O2 may also be produced directly in the liquid; we have previously analyzed the reaction channels that may be possible considering a range of reports in the literature [24]. One of the more likely reaction paths is dissociative electron attachment shown by reaction (3), followed by [24]:
A third possible reaction leading to H2O2 is the Clarke-type reaction [41] (O2 + 2H+ + 2e– → H2O2) which could occur at the anode when a large amount of O2 and H+ are present. However, we do not believe this mechanism is likely since H2O2 was also formed when we used a different plasma configuration without the anode in contact with the liquid [42]. H2O2 formation is also possible via recombination of hydroperoxyl (HO2) radicals which are generated by the following reactions (in liquid) [24, 43]:
However, reaction (8) is highly improbable since O2 is produced locally at the anode and immediately diffuses into the gas-phase, while hydrogen radicals are mainly produced at the plasma–liquid interface and are likely to induce reaction (5). In summary, H2O2 is most likely produced via reaction (6), either following OH formation through reaction (4), or gas-phase reaction (2). The relative importance of these two reactions, one in the gas phase and the other in the liquid phase remains debatable.
The experiments in an argon-controlled environment indicate the importance of air on plasma-induced chemistry with water. In particular, the results suggest that air may be the cause of acidification. It has been previously shown that air can lead to the formation of NO2 which is readily absorbed in water where it can reduce the pH of the solution via the formation of \({\text{NO}}_{2}^{ - }\) and \({\text{NO}}_{3}^{ - }\) ions [44, 45]. Rumbach et al. have reported that there are two types of reactions that occur in a plasma–liquid systems: charge transfer and plasma neutral reactions [44]. The decrease in pH in air experiments may be due to the plasma neutral reactions where the gaseous ions (e.g., NO2), radicals, molecules, etc. from gas-phase chemistry dissolve in the liquid to form HNO3 and HNO2 acid via the following reactions (in liquid):
and
This is confirmed by the absence of a pH change when water is treated in an argon background. The argon-controlled environment experiments also showed that the H2O2 concentration was two to three times higher than for a background of air. This may indicate that when the experiments are carried out in air, H2O2 is decomposed over time. H2O2 can be decomposed by \({\text{NO}}_{2}^{ - }\) ions through the following reaction (in liquid) [46]:
The rate of reaction (11) is very high in acidic solution and it decreases as the acidity is reduced. Therefore, under an argon atmosphere, the H2O2 concentration remains high because \({\text{NO}}_{2}^{ - }\) ions are not formed, while in air, H2O2 is still produced, but it is consumed via reaction with \({\text{NO}}_{2}^{ - }\) ions. Since the ratio of the number of electrons to that of H2O2 molecules is ~2 in the first few minutes under argon, the electrons do appear to be primarily involved in reactions involving precursors to H2O2. We emphasize that while our experiments support the qualitative nature of our mechanistic description, further work is required to evaluate the kinetics and provide a complete quantitative picture.
CONCLUSIONS
We have demonstrated that the temperature, pH, and conductivity of water are influenced by plasma treatment. We also observed the formation of molecular O2 in the gas and liquid phase (dissolved), and H2O2 in solution. Experiments conducted in argon and in open air show distinct differences that point to the role of O2 and N2 in air. The relation between the hydrogen peroxide concentrations and the current of plasma processing revealed that a major part of the injected electrons contribute to formation of H2O2 via the reactions either at the plasma-water interface or from gas-phase reactions. Based on this analysis we have produced a qualitative reaction model leading to a possible description of the plasma-water interactions in this case. The understanding of this simple plasma-induced chemistry with water can open up a vast range of plasma-activated chemistry in liquid with enormous potential for the synthesis of chemical compounds, nanomaterials synthesis and functionalization and a wide range of applications such as water treatment and the emerging field of plasma medicine [47]. In addition to the immense application potential, we would like to emphasize that there might be opportunities for dialing reactions in the liquid phase (e.g., inset of Fig. 6). Hybrid plasma-liquid models [48–50] supported by more experimental evidence will be required to give a full quantitative account of all reactions. Our contribution here has however highlighted important aspects that will be essential and will form the basis for future developments in this new scientific endeavor which has been possible only recently thanks to great progress in atmospheric pressure plasma science.
REFERENCES
F. Rezaei, P. Vanraes, A. Nikiforov, R. Morent, and N. de Geyter, Materials 12, 2751 (2019).
P. Bruggeman et al., Plasma Sources Sci. Technol. 25, 53002 (2016).
D. Mariotti, J. Patel, V. Švrček, and P. Maguire, Plasma Process. Polym. 9, 1074 (2012).
P. Bruggeman and C. Leys, J. Phys. D: Appl. Phys. 42, 053001 (2009).
D. B. Graves, J. Phys. D: Appl. Phys. 45, 263001 (2012).
Y. Yang, Y. I. Cho, and A. Fridman, Plasma Discharge in Liquid: Water Treatment and Applications (CRC, Boca Raton, FL, 2012).
D. Mariotti and R. M. Sankaran, J. Phys. D: Appl. Phys. 43, 323001 (2010).
R. Akolkar and R. M. Sankaran, J. Vac. Sci. Technol. A 31, 050811 (2013).
S. Samukawa et al., J. Phys. D: Appl. Phys. 45, 253001 (2012).
K. Oehmigen, M. Hähnel, R. Brandenburg, C. Wilke, K. D. Weltmann, and T. von Woedtke, Plasma Process. Polym. 7, 250 (2010).
P. Bruggeman, T. Verreycken, M. A. Gonzalez, J. L. Walsh, M. G. Kong, C. Leys, and D. C. Schram, J. Phys. D: Appl. Phys. 43, 124005 (2010).
S. Ikawa, K. Kitano, and S. Hamaguchi, Plasma Process. Polym. 7, 33 (2012).
M. Nakase, A. Agiral, T. Nozaki, J. Gardeniers, and K. Okazaki, in Proceedings of the 29th International Conference on Phenomena in Ionized Gases, Mexico, 2009.
C. Richmonds, M. Witzke, B. Bartling, S. W. Lee, J. Wainright, C. C. Liu, and R. M. Sankaran, J. Am. Chem. Soc. 133, 17582 (2011).
N. Shainsky, D. Dobrynin, U. Ercan, S. Joshi, H. Ji, A. Brooks, G. Fridman, Y. Cho, A. Fridman, and G. Friedman, in Proceedings of the 20th International Symposium on Plasma Chemistry ISPC-20, 2011.
E. Riché, A. Carrié, N. Andin, and S. Mabic, Am. Lab. 38 (13), 22 (2006).
G. Eisenberg, Ind. Eng. Chem., Anal. Ed. 15, 327 (1943).
P. Lukeš, PhD Thesis (Inst. of Plasma Physics, Prague, 2001).
Y. Lu, S. Xu, X. Zhong, K. Ostrikov, U. Cvelbar, and D. Mariotti, Europhys. Lett. 102, 15002 (2013).
P. Lukes, N. Aoki, E. Spetlikova, S. Hosseini, T. Sakugawa, and H. Akiyama, J. Phys. D: Appl. Phys. 46, 125202 (2013).
W. Graham and K. Stalder, J. Phys. D: Appl. Phys. 44, 174037 (2011).
S. Kanazawa et al., Plasma Sources Sci. Technol. 20, 034010 (2011).
R. Ono and T. Oda, J. Phys. D: Appl. Phys. 41, 035204 (2008).
B. R. Locke and K. Y. Shih, Plasma Sources Sci. Technol. 20, 034006 (2011).
S. K. Sengupta, R. Singh, and A. K. Srivastava, J. Electrochem. Soc. 145, 2209 (1998).
J. L. Brisset et al., Ind. Eng. Chem. Res. 47, 5761 (2008).
G. V. Buxton, C. L. Greenstock, W. P. Helman, and A. B. Ross, Phys. Chem. Ref. Data 17, 513 (1988).
L. Nemcova, F. Krcma, C. Kelsey, and W. Graham, in Proceedings of the 21st Europhysics Conference on Atomic and Molecular Physics of Ionized Gases, Portugal, 2012.
K. P. Arjunan, PhD Thesis (Drexel Univ., Philadelphia, USA, 2011).
A. Hickling and M. Ingram, Trans. Faraday Soc. 60, 783 (1964).
A. Hickling and M. Ingram, J. Electroanal. Chem. 8, 65 (1964).
S. K. Sengupta and O. P. Singh, J. Electroanal. Chem. 369, 113 (1994).
B. Abel, U. Buck, A. Sobolewski, and W. Domcke, Phys. Chem. Chem. Phys. 14, 22 (2012).
K. R. Siefermann, Y. Liu, E. Lugovoy, O. Link, M. Faubel, U. Buck, B. Winter, and B. Abel, Nat. Chem. 2, 274 (2010).
F. Uhlig, O. Marsalek, and P. Jungwirth, J. Phys. Chem. Lett. 4, 338 (2013).
Y. Muroya, S. Sanguanmith, J. Meesungnoen, M. Lin, Y. Yan, Y. Katsumura, and J. P. Jay-Gerin, Phys. Chem. Chem. Phys. 14, 14325 (2012).
B. Milosavljevic and O. Micic, J. Phys. Chem. 82, 1359 (1978).
M. Witzke, P. Rumbach, D. B. Go, and R. M. Sankaran, J. Phys. D: Appl. Phys. 45, 442001 (2012).
R. Burlica, M. J. Kirkpatrick, and B. R. Locke, J. Electrost. 64, 35 (2006).
V. I. Parvulescu, M. Magureanu, and P. Lukes, Plasma Chemistry and Catalysis in Gases and Liquids (Wiley, New York, 2012).
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications (Wiley, New York, 1980), Vol. 2.
S. Mitra, V. Švrček, D. Mariotti, T. Velusamy, K. Matsubara, and M. Kondo, Plasma Process. Polym. 11, 158 (2013).
J. Weiss, Nature (London, U.K.) 153 (7), 48 (1944).
P. Rumbach, M. Witzke, R. M. Sankaran, and D. B. Go, J. Am. Chem. Soc. 135, 16264 (2013).
P. Rumbach, M. Witzke, R. M. Sankaran, and D. B. Go, in Proceedings of the ESA Annual Meeting on Electrostatics, 2013, p. 1.
M. Anbar and H. Taube, J. Am. Chem. Soc. 76, 6243 (1954).
K. Zeng and D. Zhang, Prog. Energy Combust. Sci. 36, 307 (2010).
J. Liu et al., Sci. Rep. 6, 38454 (2016).
B. He et al., J. Phys. D: Appl. Phys. 50, 445207 (2017).
K. Tachibana and K. Yasuoka, J. Phys. D: Appl. Phys. 53, 125203 (2020).
ACKNOWLEDGMENTS
The authors are thankful to Prof. Davide Mariotti and Prof. Bill Graham for providing critical suggestions. The authors also thank Dr. Alan Brown and Mr. Paul O’Connor, Postgraduate Student at the Ulster University for helping with the oxygen measurements.
Funding
This work was supported by the Ulster University. Jenish Patel acknowledges the Ulster University Vice-Chancellor Research Studentship (VCRS) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Patel, J., Keshvani, M.J. Study of Plasma–Water Interactions: Effect of Plasma Electrons and Production of Hydrogen Peroxide. Russ. J. Phys. Chem. 95, 2691–2698 (2021). https://doi.org/10.1134/S0036024421130161
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024421130161