
Abstract
In the base catalyzed ethanol condensation reactions, the calcined MgO–Al2O3 derived hydrotalcites used broadly as catalytic material and the calcination temperature plays a big role in determining the catalytic activity. The characteristics of the hydrotalcite material treated between catalytically relevant temperatures 450 and 800 °C have been studied with respect to the physical, chemical, and structural properties and compared with catalytic activity testing. With the increasing calcination temperature, the total measured catalytic basicity dropped linearly with the calcination temperature and the total measured acidity stayed the same for all the calcination temperatures except 800 °C. However, the catalyst activity testing does not show any direct correlation between the measured catalytic basicity and the catalyst activity to the ethanol condensation reaction to form 1-butanol. The highest ethanol conversion of 44 % with 1-butanol selectivity of 50 % was achieved for the 600 °C calcined hydrotalcite material.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The projected crude oil depletion along with increased greenhouse gas emissions has created great interest in developing technologies to produce fuels and chemicals from renewable resources. Currently, ethanol generated from renewable resources accounts for about 90 % of worldwide biofuel production and global ethanol production is continuously increasing due to the recent inventions in renewable resource generation and conversion technologies. The overall ethanol production is anticipated to reach greater than 30 billion gallons in 2017 and beyond [1]. Due to the ethanol blending limitation in gasoline and advancements in conversion technologies, ethanol is expected to be in surplus in future years [2–4]. Technologies are already available for the conversion of ethanol to higher value compounds, such as hydrogen and acetaldehyde, to utilize the surplus ethanol [5, 6]. Another technology that can provide a platform for ethanol utilization is Guerbet ethanol condensation chemistry. This process converts ethanol to a versatile building block compound, 1-butanol, which can be readily converted to fuel compounds, such as jet fuel and diesel, or chemical compounds, such as solvents and flavoring agents [7, 8]. A basic oxide material, such as magnesium oxide (MgO), is used as catalyst for the ethanol condensation reaction [9]. The conversion of ethanol to 1-butanol occurs via a complex network of reaction mechanisms that require bi-functional acid–base catalytic materials for an increased activity [10, 11]. The hydrotalcite [Mg6Al2(CO3)(OH)16·4H2O] derived magnesium-alumina mixed oxide (MgO–Al2O3) catalyst is identified as one of the efficient catalytic materials for converting ethanol to butanol. Hydrotalcite is made up of anionic clays in which divalent magnesium cations (Mg2+) within brucite-like layers are replaced by aluminum trivalent cations (Al3+), resulting in positively charged layers whereby the charge is compensated by interlayer anions, in this case carbonates (CO3 2−), and by weakly bonded water [12, 13]. The catalyst synthesis procedure, treatment method, catalyst composition, and the experimental conditions play a major role in directing the ethanol condensation chemistry. The fresh hydrotalcite with carbonate as the compensating anion displays no catalytic activity in the aldol condensation reaction. After calcination, the hydrotalcites are in the form of MgO–Al2O3 mixed oxides and display substantial activity [14]. In particular, the calcination temperature affects both the physical properties and the chemical properties of the hydrotalcite derived mixed oxides. Our experimental results show a change in catalytic activity with respect to the ethanol condensation reaction. In this paper we will discuss the experimental and catalyst characterization results in detail to demonstrate the role of calcination temperature on the hydrotalcite derived MgO–Al2O3 when converting ethanol to 1-butanol.
2 Experimental
2.1 Materials and Catalyst Testing
The catalyst of interest in this work, hydrotalcite, was purchased from Sigma-Aldrich. Ethanol (200 proof) was purchased from Decon Labs, Inc. The catalyst testing was conducted on a down flow gas-phase stainless steel tube reactor arrangement. The catalyst of interest was placed in the middle of the reactor tube in an isothermal zone and heated using a small tube furnace to the desired reaction temperature. Ethanol (using a syringe pump) and carrier gas nitrogen (N2) were fed from the top of the reactor. The liquid product samples were collected in the bottom of the reactor in a cold trap (ice bath) arrangement. Later, the collected liquid products were analyzed in the gas chromatography-flame ionization detector (GC-FID). The non-condensable gases from the cold trap passed through the flow meter and were analyzed in the gas chromatography-thermal conductivity detector (GC-TCD). Before use, the hydrotalcite material was calcined at a desired temperature between 450 and 800 °C in air atmosphere. The calcined materials were then pressed and sieved in a 60–100 mesh. The experiments reported in this article were conducted at 300 °C and at atmospheric pressure with a weight hourly space velocity (WHSV) of 0.05 h−1. Both GC-FID and GC-TCD were calibrated with samples of know concentration.
2.2 Catalyst Characterization
X-ray powder diffraction (XRD) patterns of all calcined hydrotalcite samples were recorded on a Phillips X-Pert (50 kV and 40 mA) diffractometer using Cu Kα radiation (λ¼ 1.5437 Å). Each sample was scanned in the range between 20° and 80°. Surface area, pore volume, and pore size analyses were determined using N2 adsorption/desorption collected with a QUANTACHROME AUTOSORB 6-B gas sorption system. The samples were degassed at 100 °C for approximately 10 h under vacuum. An N2 adsorption/desorption at a constant temperature of 77.4 K was conducted on the degassed samples. The volume amount of N2 gas that adsorbed/desorbed onto the surface of the sample versus the relative pressure was measured (isotherm). The surface area was determined using adsorption data at p/po value between 0.05 and 0.2 using the Brunauer–Emmett–Teller (BET) method. The Barrett–Joyner–Halenda (BJH) method was used for the porosity and pore size analyses. The morphological appearance of the catalysts was observed by scanning electron microscopy (SEM) using a FEI Helios Nanolab 600 scanning electron microscope. Ammonia (NH3) and carbon dioxide (CO2) temperature programmed desorption (TPD) analyses were performed on calcined hydrotalcite catalysts. For the NH3–TPD measurement, sample material was saturated with NH3 at room temperature in a flow of 15.7 % NH3 in helium (He) and for the CO2–TPD measurement, sample material was saturated with CO2 at room temperature in a flow of 5 % CO2 in He. After saturation, the weakly bound NH3/CO2 was desorbed prior to the measurement at 50 °C for 3 h at a He flow rate of 25 ml min−1. The desorption curve was then attained at a heating ramp of 10 °C min−1 from 50 to 800 °C at a He flow rate of 25 ml min−1. The off-gas was analyzed on a Micromeretics Autochem II equipped with a PFEIFFER mass spectrometer. For the thermo gravimetric (TG) analysis, the fresh hydrotalcite was analyzed without any prior treatment. The TG experiment was carried out by heating a premeasured amount of spent catalyst from room temperature to 900 at 10 °C min−1 in a gas flow of He at 10 ml min−1. A NETZSCH thermogravimetry analyzer equipped with a PFEIFFER mass spectrometer was used for these analyses. Solid state 27Al (130.287 MHz) magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) measurements were carried out using a Varian VNMR spectrometer (11.7 T, 500 MHz) equipped with a variable-temperature double resonance probe. Approximately 20 mg of finely ground powder samples were loaded into 4.0 mm zirconia PENCIL rotors and spun at 10 kHz. 27Al MAS NMR spectra were collected at 130.287 MHz using a single-pulse excitation Bloch-decay method with a short radio frequency pulse of 0.66 μs (90° pulse width of 6.6 μs) and a repetition delay of 1 s. 27Al NMR chemical shifts were referenced to 0.1 M Al(NO3)3 at 0 ppm. A 40 ms acquisition time, 50 kHz spectral window, and 20 Hz Lorentzian line broadening were used for all the experiments.
3 Results and Discussion
Ethanol condensation (Guerbet) chemistry to generate 1-butanol occurs in a complex sequence of steps involving dehydrogenation, dehydration, aldolation, and hydrogenation reactions requiring acid–base bi-functional catalytic material for an efficient process [10]. Each of these reaction steps requires different active sites between acid and base with different strengths. Scheme 1 shows the network of reactions required to generate 1-butanol from an ethanol condensation reaction. First, ethanol dehydrogenates to form acetaldehyde, followed by aldol condensation to form 3-hydroxybutanal, then crotonaldehyde, and finally, successive hydrogenation yielding 1-butanol. The main compounds identified in the product stream of the ethanol condensation are acetaldehyde and 1-butanol. The intermediate compounds, 3-hydroxybutanol and crotonaldehyde, are very short lived and are not detected in the product composition. The main undesired by-products in this process are the ethanol dehydration compounds ethylene and diethyl ether, which run parallel to the condensation reaction sequence. Due to the cross condensation reaction between the feed ethanol and product 1-butanol as well as the direct condensation reaction between product 1-butanol(s), the product distribution also contains alcohols with a carbon number higher than C4.

Sequence of reaction mechanism for the ethanol condensation (Guerbet chemistry) to form 1-butanol
The fresh hydrotalcite (without any thermal treatment) had no catalytic activity for any base catalyzed condensation reactions [15]. The thermal treatment is crucial to convert the layered double hydroxide hydrotalcite via dehydration, dehydroxylation, and decarbonization to MgO–Al2O3 having strong Lewis basic sites associated with the Mn+O2− acid–base pair sites [16–18]. The thermal treatment of hydrotalcites at high temperatures yields homogeneously interdispersed mixed MgO–Al2O3 oxides. The calcined hydrotalcites generate strong basicity similar to the pure oxides and contain more surface defects, resulting in the incorporation of the Al3+ cations in the MgO framework and the acidic functionality due to the presence of the Al2O3 [16, 19]. Figure 1 shows the weight loss of the hydrotalcite between 100 and 900 °C. The total weight loss of ~44 % occurred in this temperature range, with the weight loss occurring in two noticeable steps. In the first step, weakly bound water is desorbed up to 200 °C. When temperature ranges between 250 and 550 °C, water and CO2 are formed via the dehydroxylation of OH groups and via decarboxylation of CO3 2− anions in the brucite layer. All of the weight loss occurred before ~550 °C; during the weight loss, the primary compounds detected were water and CO2. The release of these gaseous compounds generates a substantial porosity in the calcined material and a significant increase in the surface area of the material [19].

TG analysis of the fresh hydrotalcite samples
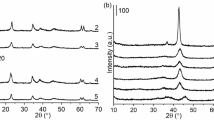
The physical properties of the catalyst tested at different calcination temperature are listed in Table 1. The BET surface areas for all the samples obtained between 450 and 700 °C are ~200 m2 g−1 and the surface area values between these samples deviates with in the ±5 %. Compared to the other samples, the sample calcined at 800 °C had a significant drop in the surface area to ~165 m2 g−1. In Fig. 2, the XRD patterns of the hydrotalcite samples calcined at different temperature is shown. For all the samples calcined between 450 and 800 °C, the reflections detected at 2θ angles around 43° and 63° resemble a MgO-like phase [20]. The peaks representing Al2O3 are very hard to identify in the XRD analysis, which indicates a very high dispersion of Al2O3 in the MgO structures and no segregation of Al2O3 for the range of calcination temperature tested.

XRD of the catalysts tested at different calcination temperatures
The diffraction peaks for the calcined hydrotalcites are narrower at higher temperatures (450–800 °C). This might indicate improved crystallinity and/or increased particle size [14] but the SEM images (see Fig. 4) indicate there is no identifiable difference between these samples. For a calcination temperature of 700 °C and beyond, a doublet peak starts to appear at ~37°, but even at 800 °C, the major phase is MgO-like. Hibino et al. reported that at 900 °C and above, the XRD reflections for the MgO–Al2O3 double oxide decomposed to spinel (MgA1204) and MgO was identified [21]. NMR is less affected by the crystallinity and the particle size and is sensitive to the phase transformations [22]. NMR can be a helpful tool to identify the changes in Al3+ cations that cannot be identified by XRD [17]. In the fresh hydrotalcite, Al3+ occupies the octahedral sites. After calcination at higher temperatures, Al3+ sites potentially change phases and start to occupy both octahedral and tetrahedral sites [17, 23]. For Al2O3, the resonance peak at a chemical shift ~1.6 ppm represents the octahedral coordination of the Al3+ and the broad small resonance peak at ~64 ppm represents the tetrahedral coordination of the Al3+ [24].
The 27Al MAS NMR for the hydrotalcite samples heated at different temperatures is shown in Fig. 3. For the samples calcined between 450 and 800 °C, a sharp resonance peak between ~+10 and −10 ppm representing the octahedral and a broad resonance peak between ~50 and 70 ppm representing the tetrahedral was identified. The peak intensities for both coordination of Al between the calcined samples (barring experimental errors) are the same. It is also reported elsewhere that the tetrahedral coordination starts at a calcination temperature of roughly ~300 °C and peaks at 400 °C. The ratio between the octahedral and tetrahedral stays relatively constant around 1.4 between the temperatures of our interest; it is reported in the literature that the ratio starts to change beyond 900 °C [23] with the predominance of Al3+ coordination in the octahedral position. At higher temperatures, the Al3+ cations convert from octahedral coordination to tetrahedral coordination via isomorphs substitution of Mg2+ [25]. The newly created positive charge can be compensated by the generation of different defects such as cationic vacancies [14]. The nature of the MgO rich hydrotalcite “memory effect” to reconstruct back to the parent hydrotalcite when exposed to moisture reduces the chances of bulk Al2O3 segregation and maintains both the tetrahedral and octahedral coordinated Al3+ to the MgO matrix [25, 26].

1–D 27Al MAS NMR spectra of hydrotalcite samples treated between 450 and 800 °C in air
SEM imaging was used to study the morphology and the particle size distributions of the calcined hydrotalcite samples at different temperatures. The SEM images of the samples calcined between 450 and 800 °C are shown in the Fig. 4. As reported by Erickson et al. the structure is crystalline after the high temperature thermal treatment and forms a hexagonal structure. The particle size for the catalysts was similar between the calcination temperatures. The typical individual particle size was in the range between 500 nm and 1 µ. At the higher temperatures, the mixed oxide crystals start to cluster together with some individual crystals [27]. At 800 °C, clustering is at higher level, which can be related to the lower surface area from the BET analysis for this sample compared to the other calcined samples. The individual particle agglomerates were crushed to disperse on a Silcon wafer for the imaging; hence this clustering is not directly evident looking at the SEM image of 800 °C sample. Overall, the flat surface is retained with varying cluster size with respect to the calcination temperature. In general, the activation of Mg–Al hydrotalcites via thermal treatment is considered to involve the replacement of interlayer CO3 2− by hydroxyl groups without any extreme effect on the original ordered stacked structure [14].

SEM images of the hydrotalcite material calcined at different temperatures
The catalytic base site densities and strengths were obtained from the CO2 TPD analysis. The CO2 TPD profiles of the hydrotalcite materials treated at different calcination temperatures as a function of CO2 desorption temperature are displayed in Fig. 5. The TPD CO2 profile shows two distinct peaks. The first peak was between 75 and 150 °C (identified as low temperature peak) and the second peak was between 150 and 400 °C (identified as high temperature peak) based on the calcination temperature of the sample material. Between the calcination temperatures there is very minimal change in the low temperature CO2 desorption peak. Significant changes with respect to the peak intensity and the CO2 desorption temperature is identified in the high temperature peak. The peak shape and sizes are similar for the 450 and 550 °C calcined samples and for the 600, 700, and 800 °C calcined samples.

CO2 TPD profiles of the hydrotalcite materials treated at different calcination temperature
Table 2 contains the base site densities based on the temperature at which CO2 desorbed in the TPD experiment. The amount of desorbed CO2 at different temperature regions was measured by the TPD curve integration. Based on the CO2 TPD profile, it can be concluded that the calcined hydrotalcites contains more than one type of basic sites with different strengths and densities. The desorbed CO2 at the low temperature region shows a bell curve like trend with a peak at 600 °C. The desorbed CO2 at the high temperature region shows a linearly dropping trend with an increase in the catalyst calcination temperature. Shen et al. reported that for the Mg rich MgO–Al2O3, Lewis acidity is attributed to the surface defects with accessible Al3+ cations in Al3+–O2−–Mg2+ species and density of the Lewis acidity is orders of magnitude higher in comparison to the Brønsted acidity for the MgO–Al2O3, with three to one ratio composition [17, 26]. The catalytic acid site densities and the strengths were obtained from the NH3 TPD analysis. The NH3 TPD profiles of the hydrotalcite materials treated at different calcination temperatures as a function of desorption temperature is shown in Fig. 6. The NH3 profile contains two overlapping curves for all the calcination temperature materials tested. Even though the curve shape looks dissimilar between the different calcination temperatures, the area under the curve is very similar with the exception of the 800 °C calcination temperature.

NH3 TPD profiles of the hydrotalcite materials treated at different calcination temperatures
Figure 7 compares the ethanol conversion rate with respect to the total basic sites determined by the CO2 adsorption from TPD analysis. The result shows that the ethanol conversion rate does not have any direct correlation with the total number of basic sites identified. Comparing to the base sites calculated from the low temperature region (see Table 2) the ethanol conversion rates have a direct relationship to the weakly desorbed sites, but the difference between the values (low temperature region) compared to the total number of sites calculated is too small. Therefore, making conclusions regarding the active sites nature based on the CO2 desorption numbers alone becomes difficult.

Ethanol conversion rate as a function of the number of base sites determined by the CO2 adsorption for the catalyst material derived from hydrotalcite at different calcination temperatures
As mentioned previously, an ethanol condensation reaction on hydrotalcite derived MgO–Al2O3 occurs via a complex network of reactions occurs on bi-functional acid–base catalysis that contains acidic and basic sites with varying strength and nature. The influence of hydrotalcite derived MgO–Al2O3 catalytic activity towards the generation of dehydration products from ethanol with respect to the catalyst calcination temperature is shown in Fig. 8. The overall mass balance of these experiments was between 97 and 100 %. The selectivity to ethylene stays constant between 4 and 5 % for all the calcination temperatures, but the diethyl ether selectivity drops continuously with respect to the increasing catalyst calcination temperature. The highest selectivity of ~36 % observed for the 450 °C calcination temperature and the lowest selectivity ~24 % observed for the 800 °C calcination temperature.

Ethanol conversion and the carbon selectivity of the dehydration products for the different calcination temperatures of the catalyst
Over acidic sites, ethanol goes thru intramolecular dehydration to form ethylene and intermolecular dehydration to form diethyl ether; both dehydration compounds are thermodynamically favorable with respect to the catalyst acid strength and the operating temperature regime [28]. Cosimo et al. conducted an experiment by varying the Mg to Al ratio in the catalyst composition and reported that the ethylene and diethyl ether rate increased with Al content due to the density increase of both Al3+–O2− pairs and low and medium strength basic sites [19]. Unlike ethylene, the formation of the diethyl ether requires two alcohol molecules adsorbed on two different active sites and one of the responsible sites may belong to the strong basic strength (drops continuously with the increasing calcination temperature; see Table 2). This may be the reason for the lowering production rate of diethyl ether with an increasing catalyst calcination temperature. The influence of hydrotalcite derived MgO–Al2O3 catalytic activity towards the generation of Guerbet condensation products from ethanol with respect to the catalyst calcination temperature is shown in Fig. 9. The ethanol dehydrogenation product acetaldehyde selectivity stayed at ~3 % for all the calcination temperatures except for the 800 °C calcination temperature, for which the selectivity reached around 5 %. Both the 1-butanol generation and the C6+ alcohol generation showed a similar trend as the ethanol conversion and showed peaks at a calcination temperature of 600 °C. Even though there is a noticeable change in the higher alcohol selectivity, it is small compared to the change in the ethanol conversion between the lowest and the highest value (around 18 %).

Ethanol conversion and the carbon selectivity of the Guerbet condensation products for the different calcination temperatures of the catalyst
The catalytic performance of the hydrotalcite derived MgO–Al2O3 catalysts seems to be not only correlated to the density and strength of acid–base active sites, but also to the nature of the bulk structure and changes in the Mg2+ or Al3+ cation environment due to the change in treatment conditions and chemical composition [26]. This also means for the cascade chemistry such as ethanol condensation with several competing reactions requires different active sites with different strength for different reactions. Therefore, the relative populations of the active sites of different natures should give different selectivity to the products. Rao et al. reported that a catalyst generated at a calcination temperature of 475 °C performed the best for the aldol condensation between benzaldehyde and acetone [29]. Constantino et al. reported the catalyst treated at calcination temperature of ~250 °C is active [30]. By understanding the exact requirement and the nature of the active sites, an opportunity will be created to develop an active catalytic material. To increase the basic sites Kozlowski et al. added sodium (Na) to the zirconium oxide (ZrO) catalyst for the ethanol condensation reaction and reported that the production rate of acetaldehyde condensation products were not increased over the Na loaded catalyst. They suggested that the coupling of ethanol could be dominated by a large surface coverage of ethoxide (an intermediate in ethanol dehydrogenation), analogous to that observed the ethanol coupling over MgO, and concluded that the coverage of ethanol plays a major role in the 1-butanol production rate from ethanol. Trends based on base site density and strength can be deceptive, since the strongly bonded ethoxide may decrease the available base sites for the condensation step [10, 31, 32]. Our experimental result shows, the highest activity for ethanol condensation chemistry with respect ethanol conversion and selectivity to 1-butanol and C6+ alcohols were observed for the catalyst calcination temperature 600 °C and the activity is lower for either side of the 600 °C calcination temperature. Therefore, we can conclude that strong basic sites and density seem to be not essential for the ethanol condensation chemistry, but require a unique combination of acid and base catalytic site at different strengths.
4 Conclusions
The MgO–Al2O3 mixed oxides derived from the hydrotalcite showed good promise for synthesizing 1-butanol from ethanol in one catalytic step. Calcination temperature played a role in modifying the chemical properties of the hydrotalcite derived MgO–Al2O3 catalyst as well as in the catalyst activity for the 1-butanol synthesis. Over hydrotalcites calcination temperature between 450 and 700 °C had a very minimal effect on the physical property such as surface area but had a huge effect on the basic site strength and density. Based on the CO2 and NH3 TPD analysis, it was found that basicity of the catalytic material is the decisive element in the performance of the ethanol condensation chemistry and at the same time, presence of the adjacent acidic sites with optimum balance is also important for the efficient conversion process.
References
Sun J, Wang Y (2014) Recent advances in catalytic conversion of ethanol to chemicals. ACS Catal 4(4):1078–1090. doi:10.1021/cs4011343
Tews IJ, Jones SB, Santosa DM, Dai Z, Ramasamy K, Zhu Y (2010) A survey of opportunities for microbial conversion of biomass to hydrocarbon compatible fuels. vol PNNL-19704. PNNL, Richland
Ramasamy KK, Wang Y (2014) Ethanol conversion to hydrocarbons on HZSM-5: effect of reaction conditions and Si/Al ratio on the product distributions. Catal Today 237:89–99. doi:10.1016/j.cattod.2014.02.044
Ramasamy KK, Zhang H, Sun JM, Wang Y (2014) Conversion of ethanol to hydrocarbons on hierarchical HZSM-5 zeolites. Catal Today 238:103–110. doi:10.1016/j.cattod.2014.01.037
Ni M, Leung DYC, Leung MKH (2007) A review on reforming bio-ethanol for hydrogen production. Int J Hydrog Energy 32(15):3238–3247. doi:10.1016/j.ijhydene.2007.04.038
Angelici C, Weckhuysen BM, Bruijnincx PC (2013) Chemocatalytic conversion of ethanol into butadiene and other bulk chemicals. Chem Sus Chem 6(9):1595–1614. doi:10.1002/cssc.201300214
Ramasamy KK, Wang Y (2013) Thermochemical conversion fermentation-derived oxygenates to fuels. In: Zhang B, Wang Y (eds) Biomass processing, conversion and biorefinery. Nova Science Publishers Inc, New York, pp 289–300
Zheng J, Tashiro Y, Wang Q, Sonomoto K (2015) Recent advances to improve fermentative butanol production: genetic engineering and fermentation technology. J Biosci Bioeng 119(1):1–9. doi:10.1016/j.jbiosc.2014.05.023
Ndou AS, Plint N, Coville NJ (2003) Dimerisation of ethanol to butanol over solid-base catalysts. Appl Catal A 251(2):337–345. doi:10.1016/s0926-860x(03)00363-6
Kozlowski JT, Davis RJ (2013) Heterogeneous catalysts for the Guerbet coupling of alcohols. ACS Catal 3(7):1588–1600. doi:10.1021/cs400292f
Carvalho DL, Borges LEP, Appel LG, Ramírez de la Piscina P, Homs N (2013) In situ infrared spectroscopic study of the reaction pathway of the direct synthesis of n-butanol from ethanol over MgAl mixed-oxide catalysts. Catal Today 213:115–121. doi:10.1016/j.cattod.2013.03.034
Debecker DP, Gaigneaux EM, Busca G (2009) Exploring, tuning, and exploiting the basicity of hydrotalcites for applications in heterogeneous catalysis. Chemistry 15(16):3920–3935. doi:10.1002/chem.200900060
Roelofs JCAA, Bokhoven JAV, Dillen AJV, John WG, Jong KPD (2002) The thermal decomposition of Mg ± Al hydrotalcites: effects of interlayer anions and characteristics of the final structure. Chem Eur J 8(24):5571–5579
Xie W, Peng H, Chen L (2006) Calcined Mg–Al hydrotalcites as solid base catalysts for methanolysis of soybean oil. J Mol Catal A: Chem 246(1–2):24–32. doi:10.1016/j.molcata.2005.10.008
Chimentao R, Abello S, Medina F, Llorca J, Sueiras J, Cesteros Y, Salagre P (2007) Defect-induced strategies for the creation of highly active hydrotalcites in base-catalyzed reactions. J Catal 252(2):249–257. doi:10.1016/j.jcat.2007.09.015
Liu Y, Lotero E, Goodwin JG, Mo X (2007) Transesterification of poultry fat with methanol using Mg–Al hydrotalcite derived catalysts. Appl Catal A 331:138–148. doi:10.1016/j.apcata.2007.07.038
Shen JY, Tu M, Hu C (1998) Structural and surface acid/base properties of hydrotalcite-derived MgAlO oxides calcined at varying temperatures. J Solid State Chem 137(2):295–301. doi:10.1006/jssc.1997.7739
Rey F, Fornes V, Rojo JM (1992) Thermal-decomposition of hydrotalcites—an infrared and nuclear-magnetic-resonance spectroscopic study. J Chem Soc Faraday Trans 88(15):2233–2238. doi:10.1039/Ft9928802233
Cosimo JID, D´ıez VK, Xu M, Iglesia E, Apesteguia CR (1998) Structure and surface and catalytic properties of Mg-Al basic oxides. J Catal 178:499–510
Kuśtrowski P, Chmielarz L, Bożek E, Sawalha M, Roessner F (2004) Acidity and basicity of hydrotalcite derived mixed Mg–Al oxides studied by test reaction of MBOH conversion and temperature programmed desorption of NH3 and CO2. Mater Res Bull 39(2):263–281. doi:10.1016/j.materresbull.2003.09.032
Hibino T, Tsunashima A (1997) Formation of spinel from a hydrotalcite-like compound at low temperature: reaction between edges of crystallites. Clays Clay Miner 45(6):842–853. doi:10.1346/Ccmn.1997.0450608
Akitt JW (1989) Multinuclear studies of aluminum compounds. Prog Nucl Magn Reson Spectrosc 21:1–149. doi:10.1016/0079-6565(89)80001-9
MacKenzie KJD, Meinhold RH, Sherriff BL, Xu Z (1993) 27Al and 25 Mg solid-state magic-angle spinning nuclear magnetic resonance study of hydrotalcite and its thermal decomposition sequence. J Mater Chem 3(12):1263–1269
Park T-J, Choi S-S, Kim Y (2009) 27Al solid-state NMR structural studies of hydrotalcite compounds calcined at different temperatures. Bull Korean Chem Soc 30(1):149–152
Corma A, Fornes V, Rey F (1994) Hydrotalcites as base catalysts—influence of the chemical-composition and synthesis conditions on the dehydrogenation of isopropanol. J Catal 148(1):205–212. doi:10.1006/jcat.1994.1202
Díez V (2003) Effect of the chemical composition on the catalytic performance of MgyAlOx catalysts for alcohol elimination reactions. J Catal 215(2):220–233. doi:10.1016/s0021-9517(03)00010-1
Erickson KL, Bostrom TE, Frost RL (2005) A study of structural memory effects in synthetic hydrotalcites using environmental SEM. Mater Lett 59(2–3):226–229. doi:10.1016/j.matlet.2004.08.035
Ramasamy KK, Gerber MA, Flake M, Zhang H, Wang Y (2014) Conversion of biomass-derived small oxygenates over HZSM-5 and its deactivation mechanism. Green Chem 16(2):748–760. doi:10.1039/C3gc41369a
Rao KK, Gravelle M, Valente JS, Fc Figueras (1998) Activation of Mg–Al hydrotalcite catalysts for aldol condensation reactions. J Catal 173:115–121
Constantino VRL, Pinnavaia TJ (1994) Structure-reactivity relationships for basic catalysts derived from a Mg2+/A13+/CO layered double hydroxide. Catal Lett 23:361–367
Kozlowski JT, Davis RJ (2013) Sodium modification of zirconia catalysts for ethanol coupling to 1-butanol. J Energy Chem 22(1):58–64. doi:10.1016/s2095-4956(13)60007-8
Birky TW, Kozlowski JT, Davis RJ (2013) Isotopic transient analysis of the ethanol coupling reaction over magnesia. J Catal 298:130–137. doi:10.1016/j.jcat.2012.11.014
Acknowledgments
The Pacific Northwest National Laboratory is operated by the Battelle Memorial Institute for the U.S. Department of Energy under Contract No. DE-AC05-76RL01830. This work was supported by the U.S. Department of Energy’s Bioenergy Technology Office. The SEM imaging portion of the work was done as a part of chemical imaging initiative, a laboratory directed research and development program at Pacific Northwest National Laboratory. The SEM imaging was conducted in the William R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by DOE’s Office of Biological and Environmental Research and located at PNNL. The authors wish to express thanks to Robert A. Dagle and Michael A. Lilga for the valuable technical discussions, Colin D. Smith for the XRD analysis, and Satish Nune for the TG analysis.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Ramasamy, K.K., Gray, M., Job, H. et al. Role of Calcination Temperature on the Hydrotalcite Derived MgO–Al2O3 in Converting Ethanol to Butanol. Top Catal 59, 46–54 (2016). https://doi.org/10.1007/s11244-015-0504-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-015-0504-8