
Abstract
A series of nickel complexes [Ni(L1)Br2] (C1), [Ni(L2)Br2] (C2) and [Ni(L3)Br2] (C3) (L1 = N-isopropyl-N-(((diphenylphosphanyl)methyl)dimethylsilyl)-1,1-diphenylphosphanamine, L2 = N-cyclopentyl-N-(((diphenylphosphanyl)methyl)dimethylsilyl)-1,1-diphenylphosphanamine, L3 = N-(2,6-diisopropylphenyl)-N-(((diphenylphosphanyl)methyl)dimethylsilyl)-1,1-diphenylphosphanamine) were synthesized and characterized by elemental analysis, mass spectrometry, spectroscopy and single-crystal X-ray diffraction. L1, L2 and L3 each act as bidentate ligands. Upon activation with ethylaluminum dichloride, these complexes produce efficient catalytic systems for selective dimerization of ethylene to 1-butene, with catalytic activities of 3.45 × 107 g/(molNi·h) and 95.6% butene selectivity containing 87.6% 1-butene fraction.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ethylene oligomerization is an important method for the production of α-olefins [1]. In the past two decades, nickel complexes have emerged as transition metal catalysts for ethylene oligomerization and have attracted wide attention in both academic and industrial research [2,3,4,5,6]. The traditional nickel catalysts mainly produce a wide Schulz–Flory (S–F) distribution which contains a small proportion of each α-olefin fraction, particularly 1-butene. The low selectivity for 1-butene has been mainly attributed to the high isomerization rate of nickel catalysts during ethylene oligomerization [7]. 1-Butene is used as a co-monomer in the production of high-density polyethylene (HDPE) and linear low-density polyethylene (LLDPE). Therefore, the production of 1-butene directly influences the market for both HDPE and LLDPE. The global annual demand for 1-butene was reported as 1.6 million metric tons in 2011 and is estimated to reach up to 3.5 million metric tons in 2025 [2]. The low rate of production and rapid increase in demand have stimulated research into nickel-based catalysts to meet these challenges.
The structure of the ligand plays a crucial role in controlling the catalytic performance of a metal complex, especially the product selectivity in ethylene oligomerization. In order to develop more efficient catalytic systems, extensive research has been focused on the design of new ligands. Nickel complexes with different donor ligands, including NN, NO, NP and PO donors, have been investigated for selective ethylene oligomerization [3]. The chelating NO donor (imino/amino)phenol nickel(II) complexes reported by Ngcobo et al. [4] demonstrated that the activity and selectivity of these complexes for 1-butene show opposite trends to each other, such that increasing selectivity can diminish activity, and vice versa. Campora and coworkers [8] demonstrated that the product selectivity toward 1-butene of nickel(II) complexes with NP donor phosphinito-imine ligands was also influenced by the steric bulk and showed different selectivities when different substituents were attached to the P atom. Recently, we reported nickel(II) complexes based on PNSiP ligands, which proved to be efficient for the catalytic production of 1-butene [9]. Based on our previous findings, we aimed to design and investigate nickel(II) complexes with silicon-bridged diphosphinoamine ligands (PNSiCP) for selective ethylene oligomerization of 1-butene. The effects of the reaction conditions on the activity and product selectivity of these catalysts were investigated. Our results are reported in this paper.
Experimental
Materials and methods
All the proligands and their complexes were synthesized under a nitrogen atmosphere, in a glovebox or in oven-dried flasks using standard Schlenk techniques. Bis(phenyl)phosphorus chloride, diphenylphosphine, dichlorodimethylsilane, n-butyllithium (2.4 mol/L in n-hexane) and NiBr2(DME) were purchased from Aldrich and were used as received. Lithium diphenylphosphine was prepared using the method reported by Peterson [10]. Lithium (diphenylphosphanyl)(alkyl/aryl)amide and chlorodimethyl ((diphenylphosphino) methyl) silane were prepared using the method reported by Zhang [11, 12]. Ethylaluminum dichloride (EADC, 0.9 mol/L in heptane), diethylaluminum chloride (DEAC, 2.0 mol/L in hexane), methylaluminoxane (MAO, 1.4 mol/L in toluene) and triethylaluminum (TEA) were purchased from Albemarle Corp (USA). Dried methylaluminoxane (DMAO) was prepared by pumping off all the volatile compounds from MAO at 40 °C for 6 h. Methylcyclohexane, dichloromethane, triethylamine and n-hexane were dried, degassed and purified by distillation prior to use. Polymerization-grade ethylene was purchased from Tianjin Summit Specialty Gases (China). NMR spectra were obtained with a Bruker AVANCEIII 400 M spectrometer (400 MHz for 1H, 162 MHz for 31P and 100 MHz for 13C) at 298 K in non-aqueous solvent with tetramethylsilane (TMS) as an internal reference. Elemental analyses were obtained using an Elemental Vario EL analyzer. Mass spectra were recorded using a MALDI-TOF mass spectrometer at the Chinese Academy of Sciences (Shanghai Institute of organic chemistry). GC was performed using an Agilent 7890A GC chromatograph with a flame ionization detector (FID) and an HP-5 GC capillary column (length 60 m; diameter 0.32 mm; film thickness 0.25 μm).
Synthesis of organic precursors
The proligands L1–L3 were prepared as shown in Scheme 1. A solution of CH3MgI in Et2O (20 mL) was added dropwise at − 35 °C to a solution of chlorodipenylphosphane (29.8 g, 135.06 mmol) in Et2O (100 mL) at − 35 °C under N2 and the mixture was stirred for overnight. After filtration, the organic phase was washed with distilled water (3 × 10 mL), separated and dried with anhydrous Na2SO4. Methyldiphenylphosphine (20 g, 99.90 mmol) was obtained as a colorless liquid by reduced pressure distillation in 74% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.6 (d, 3H, CH3), 7.3–7.7 (m, 10H, Ph). 31P NMR (162 MHz, CDCl3, 298 K): δ − 26.8 (s).

Synthesis and structures of the ligands. L1 R = isopropyl L2 R = cyclopentyl L3 R = 2,6-diisopropylphenyl
A solution of n-BuLi (21.5 mL, 2.4 mol/L in n-hexane, 51.6 mmol) in n-hexane (30 mL) was added dropwise to a solution of methyldiphenylphosphine (10 g, 49.95 mmol) and TMEDA (6.1 g, 25 mL) in n-hexane (15 mL) at below 0 °C. The reaction mixture was then stirred for 60 h under N2 at room temperature. The yellow precipitate was filtered off and washed with n-hexane (3 × 20 mL) to give (diphenylphosphino)methyllithium-TMEDA (16 g, 49.6 mmol, 99% yield).
A solution of (diphenylphosphino)methyllithium-TMEDA complex (16 g, 49.6 mmol) in n-hexane (50 mL) was added to a stirred solution of dichlorodimethylsilane (12.8 g, 99.17 mmol) in n-hexane (25 mL) at − 35 °C. The mixture was stirred overnight at room temperature. The precipitated LiCl was filtered out, and a yellowish oily semisolid product was obtained which still contained TMEDA. Distillation in a vacuum gave a pure colorless oil of ((chlorodimethylsilyl)methyl)diphenylphosphane (8 g) in 57% yield. 1H NMR (400 MHz, C6D6, 298 K): δ 0.00 (s, 6H, SiMe2), 1.38 (s, 2H, CH2), 6.87–7.21 (m, 10H, Ph). 31P NMR (162 MHz, C6D6, 298 K): δ − 24.45 (s).
Synthesis of L1
A solution of isopropylamine (1.9 mL, 22.22 mmol) in tetrahydrofuran (10 mL) was added at − 35 °C to a solution of Et3N (4.6 mL, 3.37 g, 33.33 mmol) in tetrahydrofuran (10 mL). A solution of chlorodipenylphosphane (2.0 mL, 2.45 g, 11.11 mmol) in tetrahydrofuran (10 mL) was then added. The mixture was stirred overnight at room temperature. The precipitated quaternary ammonium salt was filtered out, and the pale yellow filtrate was concentrated to give a yellow residue, which was recrystallized from hexane to give N-isopropyl-1,1-diphenylphosphanamine (L1a) (1.37 g, 5.64 mmol) in 51% yield. A solution of n-BuLi (2.4 mL, 2.4 mol/L in n-hexane, 5.64 mmol) was added dropwise at − 35 °C to a solution of L1a (1.37 g, 5.64 mmol) in n-hexane (10 mL). The mixture was stirred at room temperature for 5 h. After filtering and washing with n-hexane (2 × 10 mL) and drying under vacuum, the solid lithium (diphenylphosphanyl)(isopropyl)amide (L1b) (0.7256 g, 2.91 mmol, 51% yield) was obtained.
A solution of L1b (0.7256 g, 2.91 mmol) in n-hexane (5 mL) at − 35 °C was added dropwise to a solution of ((chlorodimethylsilyl)methyl)diphenylphosphane (0.8521 g, 2.91 mmol) in n-hexane (10 mL) under stirring. The mixture was stirred overnight at room temperature and then filtered. The filtrate was concentrated under vacuum, followed by washing with n-hexane (3 × 10 mL), and then dried again in a vacuum. The white solid ligand L1 was collected after recrystallization from hexane at − 35 °C and drying in a vacuum (71% yield). 1H NMR (400 MHz, C6D6, 298 K): δ 0.15 (s, 6H, SiMe2), 0.96–0.98 (d, 6H, CH3), 1.54 (s, 2H, CH2), 3.19–3.26 (m, 1H, CH), 7.02–7.48 (m, 20H, Ph). 31P NMR (162 MHz, C6D6, 298 K): δ-24.49 (s, C–P), 34.80 (s, N–P). 13C NMR (100 MHz, C6D6, 298 K): δ 0.18, 0.22, 15.50, 15.82, 23.42, 23.49, 45.81, 46.14, 125.73, 125.78, 125.91, 125.98, 126.13, 128.71, 128.91, 128.99, 129.71, 129.90, 129.99, 130.11, 130.22, 130.31, 130.42, 131.97, 132.09, 132.22, 137.91, 138.06, 141.05, 141.19.
Synthesis of L2
The proligand L2 was prepared by the same method as described for L1. Cyclopentylamine (2.2 mL, 1.89 g, 22.22 mmol), Et3N (4.6 mL, 3.37 g, 33.33 mmol) and chlorodipenylphosphane (2 mL, 2.45 g, 11.11 mmol) were used to prepare the intermediate compound N-cyclopentyl-1,1-diphenylphosphanamine (L2a) (1.62 g, 6.01 mmol) in 54% yield. Then, n-BuLi (2.5 mL, 2.4 mol/L in n-hexane, 6.01 mmol) was used to obtain lithium (diphenylphosphanyl)(cyclopentyl)amide (L2b) (1.24 g, 4.50 mmol) in 75% yield. Similarly, L2 (0.295 g, 0.56 mmol, 56% yield) was collected as a white solid, by reacting L2b (0.275 g, 1.00 mmol) and ((chlorodimethylsilyl)methyl)diphenylphosphane (0.293 g, 1.00 mmol). 1H NMR (400 MHz, C6D6, 298 K): δ 0.27 (s, 6H, SiMe2), 1.16–1.63 (m, 8H, CH2), 1.81 (s, 2H, Si-CH2-P), 3.58–3.67 (m, 1H, CH), 7.01–7.67 (m, 20H, Ph). 31P NMR (162 MHz, C6D6, 298 K): δ − 22.61 to − 22.58 (d, C–P), 46.60 (br, N–P). 13C NMR (100 MHz, C6D6, 298 K): δ 1.05, 2.91, 2.95, 2.99, 3.04, 17.50, 17.91, 22.68, 23.80, 34.13, 34.17, 61.32, 61.36, 128.06, 128.12, 128.17, 128.20, 128.27, 131.97, 132.17, 132.57, 132.77, 140.41, 140.59, 141.60, 141.76.
Synthesis of L3
L3 was prepared by the same method as described for L1. 2,6-Diisopropylamine (4.2 mL, 22.22 mmol), Et3N (4.6 mL, 3.37 g, 33.33 mmol) and chlorodipenylphosphane (2 mL, 2.45 g, 11.11 mmol) were used to prepare the intermediate: N-2,6-Diisopropylphenyl-1,1-diphenylphosphanamine (L3a) (2.29 g, 6.34 mmol) in 57% yield. Next, n-BuLi (2.64 mL, 2.4 mol/L in n-hexane, 6.34 mmol) was used to obtain lithium (2,6-diisopropylphenyl)(diphenylphosphanyl)amide (L3b) (2.98 g, 8.11 mmol) in 73% yield. Solid L3 (0.278 g, 0.45 mmol, 45% yield) was collected, from the reaction of L3b (0.37 g, 1.00 mmol) with ((chlorodimethylsilyl)methyl)diphenylphosphane (0.293 g, 1.00 mmol). 1H NMR (400 MHz, C6D6, 298 K): δ 0.30 (s, 6H, SiMe2), 0.65–0.66 (d, 6H, CHMe2), 1.15–1.17 (d, 6H, CHMe2), 1.89 (s, 2H, CH2), 3.48–3.55 (m, 2H, CH), 7.00–7.62 (m, 23H, Ph). 31P NMR (162 MHz, C6D6, 298 K): δ − 21.21 to − 21.18 (d, C–P), 52.89 – 52.92 (d, N–P). 13C NMR (100 MHz, C6D6, 298 K): δ 2.52, 2.57, 2.61, 3.71, 3.86, 17.67, 20.25, 22.71, 24.15, 25.18, 28.56, 124.40, 124.42, 126.17, 126.18, 128.08, 128.24, 128.31, 129.01, 132.61, 132.81, 134.67, 134.92, 139.18, 139.41, 141.50, 141.66, 142.71, 142.73, 147.65, 147.67.
Synthesis of the complexes
The nickel complexes were prepared as displayed in scheme 2.

Synthesis and structures of the precatalyst. C1 R = isopropyl C2 R = cyclopentyl C3 R = 2,6-diisopropylphenyl
Preparation of complex C1
A solution of L1 (0.0525 g, 0.105 mmol) in dichloromethane (10 mL) was added to a dichloromethane dispersion (10 mL) of NiBr2(DME) (0.0310 g, 0.1 mmol). The reaction mixture rapidly became dark brown and was stirred for 36 h at room temperature. The solvent was then evaporated under vacuum, and then the dark brown solid residue was washed three times with n-hexane. The solid was collected by filtration, and then dried in vacuum to obtain a ginger-colored nickel complex (C1). Yield 0.050 g (70%). Anal. Calcd for C30H35Br2NNiP2Si: C, 50.17; H, 4.91; N, 1.95. Found: C, 50.27; H, 5.03; N, 1.89. MALDI-TOF–MS: m/z = [M-81Br]+ = 637.0.
Preparation of complex C2
Complex C2 was prepared using the same synthetic approach as complex C1, but with L2 (0.0552 g, 0.105 mmol) and NiBr2(DME) (0.0310 g, 0.100 mmol) as starting materials. Yield 0.060 g (81%). Anal. Calcd for C32H37Br2NNiP2Si: C, 51.65; H, 5.01; N, 1.88. Found: C, 51.20; H, 4.85; N, 1.76. MALDI-TOF–MS: m/z = [M-81Br]+ = 662.9.
Preparation of complex C3
Complex C3 was prepared using the same synthetic approach as for complex C1, but with L3 (0.0650 g, 0.105 mmol) and NiBr2(DME) (0.0310 g, 0.100 mmol) as starting materials. Yield 0.0694 g (83%). Anal. Calcd for C39H45Br2NNiP2Si: C, 56.01; H, 5.42; N, 1.67. Found: C, 55.19; H, 5.27; N, 1.58. MALDI-TOF–MS: m/z = [M-81Br]+ = 755.0.
General procedure for ethylene oligomerization
Ethylene oligomerization reactions were carried out in a 150 mL glass autoclave fitted with a magnetic stirrer and temperature controller. The reactor was dried for 2 h at 105 °C prior to use and then vacuumed to cool to room temperature, then charged with high-purity nitrogen. After repeating the vacuuming and nitrogen filling operation three times, the nitrogen was replaced with ethylene. Methylcyclohexane (20 mL) was then injected into the reactor, followed by the co-catalyst. After stirring for 1 min at the required reaction temperature, the precatalyst solution was transferred into the reactor. The autoclave was immediately pressurized with 1.0 MPa of ethylene. The reaction was then run for the required time. The reactor was then quenched by cooling to 0 °C in an ice bath, and depressurized. The organic phase was analyzed with an Agilent 7890A GC system, using n-heptane as an internal standard.
Crystal structures of complexes C2 and C3
Single crystals of complexes C2 and C3 suitable for X-ray studies were obtained by two-phase diffusion of n-hexane/dichloromethane. Crystallographic data were collected on a Bruker SMART APEX CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) using the ω-scan technique. The determination of crystal class and unit cell parameters was carried out by the SMART program package. The raw frame data were processed using SAINT and SADABS [13] to yield the reflection data file. The structures were solved using the SHELXTL [14] program. Refinement was performed on F2 anisotropically for all the non-hydrogen atoms by full-matrix least-squares methods [14]. The crystallographic data and structural refinement parameters for C2 and C3 are summarized in Table 1. One dichloromethane molecule was found within the crystal structure of complex C2.
Results and discussion
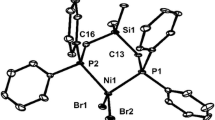
The molecular structures and selected bond parameters for complexes C2 and C3 are given in Figs. 1 and 2. Complex C2 crystallizes in the triclinic space group P-1, and complex C3 crystallizes in the monoclinic space group P2(1)/c. The nickel atom is four coordinated with a distorted square planar geometry. The coordination environment of the nickel atom for complex C3 is very similar to that of complex C2.

The molecular structure of complex C2 with 50% ellipsoids. The hydrogen atoms and dichloromethane solvent molecule are omitted for clarity. Selected bond lengths (Å) and angles (deg): Br(1)–Ni(1) = 2.3544(10), Br(2)–Ni(1) = 2.3567(8), Ni(1)–P(2) = 2.1497(13), Ni(1)–P(1) = 2.1767(12), P(1)–N(1) = 1.710(3), P(2)–C(20) = 1.808(4), Si(1)–N(1) = 1.760(4), Si(1)–C(20) = 1.889(4), N(1)–C(13) = 1.511(5); P(2)–Ni(1)–P(1) = 94.32(6), Br(1)–Ni(1)–Br(2) = 94.88(4), N(1)–P(1)–Ni(1) = 118.71(13), C(20)–P(2)–Ni(1) = 114.57(14), N(1)–Si(1)–C(20) = 106.33(18), P(1)–N(1)–Si(1) = 119.0(2), P(2)–C(20)–Si(1) = 116.7(2)

The molecular structure of complex C3 with 50% ellipsoids. The hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Br(1)–Ni(1) = 2.3330(7), Br(2)–Ni(1) = 2.3328(8), Ni(1)–P(1) = 2.1826(11), Ni(1)–P(2) = 2.2045(12), Si(1)–N(1) = 1.759(3), Si(1)–C(27) = 1.868(4), P(1)–N(1) = 1.704(3), P(2)–C(27) = 1.806(4), N(1)–C(13) = 1.465(5); P(1)–Ni(1)–P(2) = 94.64(5), Br(2)–Ni(1)–Br(1) = 91.01(3), N(1)–Si(1)–C(27) = 105.65(16), N(1)–P(1)–Ni(1) = 116.35(11), C(27)–P(2)–Ni(1) = 114.29(12), P(1)–N(1)–Si(1) = 121.10(18), P(2)–C(27)–Si(1) = 118.2(2)
After activation with EADC, the precatalysts C1-C3 were tested for selective ethylene oligomerization and showed good catalytic activity toward 1-butene and 1-hexene as well as good selectivity toward 1-butene. The insertion of 1-butene into the cationic nickel species can produce cis/trans isomers, which can decrease the selectivity of the reaction considerably. It was demonstrated that the 2,1-insertion of 1-butene followed by the β-hydride elimination produces trans-2-butene, while if Cα, Cβ rotation occurs after the 2,1-insertion followed by β-hydride elimination, the product will be cis-2-butene (Scheme 3) [15,16,17]. Similarly, 1-hexene is formed by a chain growth mechanism, while its isomers 2-hexene and 3-hexene are formed as a result of Ni-catalyzed isomerization in the same way as demonstrated for 1-butene isomers [18].

Mechanism of ethylene dimerization to butenes
Effects of co-catalyst on ethylene oligomerization
The effects of EADC, DEAC, DMAO/AlEt3 and MAO as different co-catalysts on the catalytic performance of precatalyst C3 were investigated in methylcyclohexane or toluene solvents. No activity was observed when MAO or DMAO/AlEt3 was used for activation (Table 2, entries 3 and 4), which could reflect a better stabilization of the active species with EADC and DEAC, possibly due to the chlorine atom [19]. Other activators like EADC and DEAC can successfully activate C3 precatalyst and show good selectivity toward 1-butene (Table 2, entries 1 and 2). Furthermore, in comparison with DEAC, the co-catalyst EADC showed better selectivity toward butene (Table 2, entry 1). We therefore selected EADC for further screening experiments.
Effect of C2 catalyst loading on ethylene oligomerization
The mass loading of the catalyst had a considerable influence on the catalytic performance. We found that upon decreasing the amount of precatalyst from 2.4 to 0.1 μmol, the catalytic activity and product selectivity were both significantly increased (Table 3). A very high catalytic activity (10.1 × 106 g/(molNi·h)) and good product selectivity (85.7%) were obtained toward 1-butene when the catalyst loading was reduced to 0.1 μmol (Table 3, entry 3), showing that a small amount of precatalyst could effectively suppress the formation of non-selective oligomer species [12].
Effects of N-substituent on ethylene oligomerization
To explore the influence of the N-substituent on the activity and selectivity of these catalytic systems, three catalysts were synthesized. The catalyst with the most bulky N-substituent (C3) showed highest selectivity toward 1-butene compared to the other two catalysts with smaller N-substituents (Table 4). On the other hand, the bulky substituent in C3 may hinder coordination of ethylene to the metal center, resulting in low catalytic activity.
Effects of reaction temperature on ethylene oligomerization
Our results showed that C1 and C2 displayed high selectivity at 45 °C toward 1-butene (Table 5, entries 2 and 5), while C3 showed the highest selectivity (95.6%) at 30 °C. The catalysts C1 and C2 showed high activities at the relatively low temperature of 30 °C (Table 5, entries 1 and 4). Lower activity at higher temperatures might be attributed to low ethylene solubility and/or catalyst decomposition [20]. Moreover, the low temperature could promote the formation of active species for the formation of α-olefins [21]. Compared to C1 and C2, catalyst C3 with a more bulky N-substituent showed the opposite trend toward 1-butene activity at lower temperatures.
Effects of Al/Ni molar ratios on ethylene oligomerization
In order to find the optimum Al/Ni molar ratio for these catalysts, we varied the Al/Ni molar ratios from 300 to 700 (Table 6). Catalysts C1 and C2 showed high selectivity when the Al/Ni molar ratio was 500. Upon increasing the molar ratio up to 700, the selectivity decreased, while the catalytic activity increased (Table 6, entries 1–4). On the other hand, catalyst C3 showed high selectivity as well as high activity at a relatively high (700) Al/Ni molar ratio (Table 6, entry 7).
Effect of reaction time on ethylene oligomerization
In order to investigate the effect of reaction time on the catalytic performance, we analyzed the activity and selectivity at different time intervals (10, 20, 30 and 45 min) (Table 7). The catalytic activity proved to be strongly influenced by reaction time, whereas the product selectivity remained almost unaffected. The similar selectivity toward 1-butene during the course of reaction indicates the existence of a single active species responsible for 1-butene formation, in line with previous results reported by Oliveira et al. [22]. The change in catalytic activity suggests decomposition of the active species during the course of reaction, with the formation of other species which give rise to cis/trans isomers of 1-butene [23].
Conclusion
Three nickel(II) complexes of silicon-bridged diphosphine amine ligands were synthesized, characterized and investigated as catalysts for selective ethylene dimerization. Complex C3 proved to be the most efficient catalytic system for selective ethylene dimerization, producing 88.7% of 1-C4 with 2.05 × 106 g/(molNi·h) activity under appropriate conditions, when activated by EADC co-catalyst in methylcyclohexane solvent. The steric bulk of the N-substituent was found to influence both the activity and selectivity of the catalysts. Moreover, the reaction conditions such as catalyst loading, co-catalyst type, reaction temperature, Al/Ni molar ratio and reaction time were also identified as important parameters which influence the catalytic performance.
References
Wang J, Zhang N, Li CQ, Shi WG, Lin ZY (2016) Nickel complexes based on hyperbranched salicylaldimine ligands: synthesis, characterization, and catalytic properties for ethylene oligomerization. J Organomet Chem 822:104–111
Boulens P, Pellier E, Jeanneau E, Reek JNH, Olivier-Bourbigou H, Breuil P-AR (2015) Self-assembled organometallic nickel complexes as catalysts for selective dimerization of ethylene into 1-butene. Organomet 34(7):1139–1142
Chen H-P, Liu Y-H, Peng S-M, Liu S-T (2003) New bulky phosphino–pyridine ligands. Palladium and nickel complexes for the catalytic polymerization and oligomerization of ethylene. Organomet 22(24):4893–4899
Ngcobo M, Ojwach SO (2017) Nickel(II) complexes chelated by N,N (benzimidazolylmethyl)amine ligands: synthesis and catalytic behavior in tandem ethylene oligomerization and Friedel–Crafts alkylation reactions. Inorg Chim Acta 467:400–404
Popeney CS, Rheingold AL, Guan Z (2009) Nickel(II) and palladium(II) polymerization catalysts bearing a fluorinated cyclophane ligand: stabilization of the reactive intermediate. Organomet 28(15):4452–4463
Speiser F, Braunstein P, Saussine L (2004) New nickel ethylene oligomerization catalysts bearing bidentate P,N-phosphinopyridine ligands with different substituents α to phosphorus. Organomet 23(11):2625–2632
Lecocq V, Olivier-Bourbigou H (2007) Biphasic Ni-catalyzed ethylene oligomerization in ionic liquids. Oil Gas Sci Technol 62(6):761–773
Ortiz de la Tabla L, Matas I, Palma P, Álvarez E, Cámpora J (2012) Nickel and palladium complexes with new phosphinito-imine ligands and their application as ethylene oligomerization catalysts. Organomet 31(3):1006–1016
Huang Y, Zhang L, Wei W, Alam F, Jiang T (2018) Nickel-based ethylene oligomerization catalysts supported by PNSiP ligands. Phosphorus, Sulfur Silicon Relat Elem 193(6):363–368
Peterson JD (1967) Phosphinomethyllithium compounds IV: an improved method of preparation and some synthetic applications. J Organomet Chem 8(2):199–208
Zhang L, Meng X, Chen Y, Cao C, Jiang T (2017) Chromium-based ethylene tetramerization catalysts supported by silicon-bridged diphosphine ligands: further combination of high activity and selectivity. ChemCatChem 9(1):76–79
Zhang L, Wei W, Alam F, Chen Y, Jiang T (2017) Efficient chromium-based catalysts for ethylene tri-/tetramerization switched by silicon-bridged/N,P-based ancillary ligands: a structural, catalytic and DFT study. Catal Sci Technol 7(21):5011–5018
Sheldrick GM (1996) SADABS: program for empirical absorption correction of area detector data. University of Göttingen, Göttingen
Sheldrick G (2008) A short history of SHELX. Acta Crystallogr Sect A 64(1):112–122
Dyer PW, Fawcett J, Hanton MJ (2008) Rigid N-phosphino guanidine P,N ligands and their use in nickel-catalyzed ethylene oligomerization. Organomet 27(19):5082–5087
Meng X, Zhang L, Chen Y, Jiang T (2016) Silane-bridged diphosphine ligands for nickel-catalyzed ethylene oligomerization. React Kinet Mech Catal 119(2):481–490
Sun Y, Chen Y, Mao G, Ning Y, Jiang T (2014) Boron- and silicon-bridged bis(diphenylphosphino)-type ligands for chromium-catalyzed ethylene oligomerization. Chin Sci Bull 59(21):2613–2617
Yang Q-Z, Kermagoret A, Agostinho M, Siri O, Braunstein P (2006) Nickel complexes with functional zwitterionic N,O-benzoquinonemonoimine-type ligands: syntheses, structures, and catalytic oligomerization of ethylene. Organomet 25(23):5518–5527
de Oliveira LL, Campedelli RR, Kuhn MCA, Carpentier J-F, Casagrande OL (2008) Highly selective nickel catalysts for ethylene oligomerization based on tridentate pyrazolyl ligands. J Mol Catal A: Chem 288(1):58–62
Zhang N, Wang J, Huo H, Chen L, Shi W, Li C, Wang J (2018) Iron, cobalt and nickel complexes bearing hyperbranched iminopyridyl ligands: synthesis, characterization and evaluation as ethylene oligomerization catalysts. Inorg Chim Acta 469:209–216
Aid A, Andrei RD, Amokrane S, Cammarano C, Nibou D, Hulea V (2017) Ni-exchanged cationic clays as novel heterogeneous catalysts for selective ethylene oligomerization. Appl Clay Sci 146:432–438
de Oliveira LL, da Silva SM, Casagrande ACA, Stieler R, Casagrande OL Jr (2018) Synthesis and characterization of Ni(II) complexes supported by phenoxy/naphthoxy-imine ligands with pendant N- and O-donor groups and their use in ethylene oligomerization. Appl Organomet Chem 32(7):e4414
Suo H, Zhang Y, Ma Z, Yang W, Flisak Z, Hao X, Hu X, Sun W-H (2017) 2-Chloro/phenyl-7-arylimino-6,6-dimethylcyclopenta[b]pyridylnickel chlorides: synthesis, characterization and ethylene oligomerization. Catal Commun 102:26–30
Acknowledgements
This study was supported by the National Key Research and Development Program of China (2017YFB0306700). The Natural Science Foundation of Tianjin City (14JCYBJC23100, 15JCYBJC48100 and 16JCZDJC31600).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11243_2018_276_MOESM1_ESM.doc
CCDC numbers 1859463 and 1859467 contain the supplementary crystallographic data for C2 and C3. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/. (DOC 3155 kb)
Rights and permissions
About this article

Cite this article
Wei, W., Yu, B., Alam, F. et al. Ethylene oligomerization promoted by nickel-based catalysts with silicon-bridged diphosphine amine ligands. Transit Met Chem 44, 125–133 (2019). https://doi.org/10.1007/s11243-018-0276-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0276-7