
Abstract
In this study, bis(diphenyl phosphine) dimethyl silane (L1), bis(diphenyl phosphine) methyl dimethyl silane (L2), bis(diphenyl phosphine methyl) dimethyl silane (L3) and their nickel (II) halide complexes, L1/NiBr2 (C1), L2/NiBr2 (C2), L3/NiBr2 (C3), were synthesized and characterized. The X-ray single-crystal analysis of C3 complex showed bidentate coordination on the Ni center. In combination with methylaluminoxane as co-catalyst, these complexes produced catalytic systems for ethylene dimerization to butylene with catalytic activities of up to 3.12 × 106 g mol−1 (cat.) h−1 and selectivities up to 97.6 %. With the fine-tuned ligand backbone, such a PSiP-based catalyst system provides a model for precise understanding of the impact of ligand variations on catalytic performance.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ethylene oligomerization is the primary source of α-olefins, which find application in the preparation of detergents, synthetic lubricants, plasticizers, alcohols and some copolymers [1]. Late transition metal complexes, particularly nickel-based catalysts, have served as pre-catalysts for a number of reactions, including oligomerization and polymerization of ethylene during the last decade [2–8]. These nickel catalysts have bidentate or tridentate ligands with various combinations of donor atoms such as N, P, O and S. Work on such bi-dentate nickel complexes usually include the use of N^N, N^O, N^P and P^P ligands [9]. Extensive research on ethylene polymerization using bi-dentate Ni-based catalysts bearing P^P type ligation has been reported. Wass’s group has explored the ethylene oligo-/polymerization behavior of the nickel (II) complexes. It was clear that the catalytic activity and polymer structure were very sensitive to the nature of the ligand backbone and the substituents at phosphorus. The four-membered nickel (II) chelates derived from the aminodiphosphines were very efficient catalysts for the polymerization of ethylene [10]. However, the five-membered chelates and the six-membered rings were completely inactive under the same conditions. Moreover, the larger rings were unresponsive to steric effects, and complexes of the sterically demanding ligands were inactive under the same conditions [11, 12]. The relationship between structure and activity is thus well studied for the above ligands. Relatively little, by contrast, has been reported on the application of diphosphine ligands bridging atoms (such as silicon on bridging the bridge bidentate). It will change the space structure of ligand, coordination ability and influence the central metal atoms, changing the catalytic performance [10, 13–15]. With the aim of further elucidating the role of ligand structure in these novel catalytic systems, we herein report a detailed study on the use of silicon-bridged diphosphine ligands in ethylene dimerization and oligomerization reactions [16–18]. In this paper, bis(diphenyl phosphine) dimethyl silane (L1), bis(diphenyl phosphine) methyl dimethyl silane (L2), bis(diphenyl phosphine methyl) dimethyl silane (L3) and corresponding nickel (II) halide complexes, L1/NiBr2 (C1), L2/NiBr2 (C2), L3/NiBr2 (C3), were synthesized and characterized. The effects of the reaction temperature, the molar ratio of Al/Ni and the reaction times on the catalytic activity and product selectivity were examined.
Experimental
Materials
Bis(phenyl)phosphorus chloride, diphenylphosphine, potassium hydride, dichlorodimethylsilane, chloro(chloromethyl)dimethylsilan, bis(chloromethyl)dimethylsilan, n-butyllithium (2.4 mol/L in n-hexane) and NiBr2(DME) were purchased from Aldrich and used as received. Polymerization-grade ethylene was obtained from Tianjin Summit Specialty Gases (China). Methylaluminoxane (MAO, 1.4 mol/L in toluene), toluene, dichloromethane and n-hexane were dried and degassed prior to use. 1H NMR, 13C NMR and 31P NMR spectra of the products were recorded at 25 °C on a Fourier 400 NMR spectrometer (Bruker, Billerica, MA) with tetramethylsilane (TMS) as an internal reference. All intensity data of X-ray structural determination were collected with a Bruker SMART CCD diffractometer, using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were resolved by direct methods and refined by full-matrix least squares on F2. Hydrogen atoms were considered in calculated positions. Elemental analyses were carried out on an Elemental Vario EL analyzer while mass spectra were recorded with MALDI-TOF.
Synthesis of ligands
Synthesis of bis(diphenyl phosphine) dimethyl silane (L1)
A solution of diphenylphosphine (0.76 mL, 5.00 mmol) and a suspension of KH (0.20 g, 5.00 mmol) in THF (50 mL) were stirred for 3 h at room temperature. Then the solvent was concentrated to give Ph2PK; this orange solid was added to a solution of dichlorodimethylsilane (0.27 mL, 2.25 mmol) in hexane (50 mL) and then the mixture was stirred for 12 h at 75 °C. Precipitated KCl was filtered off and the faint yellow solution was concentrated to give a yellow residue, which was recrystallized in hexane to give L1 in 80 % yield. 1H NMR (400 MHz, CDCl3) δ(ppm) 7.44 (d, J = 2.3 Hz, 8H), 7.34–7.19 (m, 12H), 0.18 (t, J = 4.0 Hz, 6H). 31P NMR (162 MHz, CDCl3) δ(ppm) −56.51(s). 13C NMR (101 MHz, CDCl3) δ(ppm) 135.40–133.37 (12C, ortho/ipso-Cquat PPh2), 129.31–127.24 (12C, meta/para-CHPPh2), −3.03 (J = 9.8 Hz, 2C, CH3).
Synthesis of bis(diphenyl phosphine) methyl dimethyl silane (L2)
2.4 M n-BuLi solution in hexanes (41.6 mL, 99.84 mmol) was added dropwise to a solution of diphenylphosphine (18.62 g, 0.1 mol) in THF (200 mL) at −80 °C. The mixture was stirred for 1 h maintaining −80 °C and subsequently stirred for 1 h at room temperature. The solvent was drained in the vacuum and n-hexane (100 mL) was added simultaneously, fully mixed dispersion after filtration. The filtrate was evaporated to afford diphenyl phosphine lithium base (18.82 g, 0.098 mol). Yield = 18.82 g (0.098 mol, 98.6 %).
A solution of diphenyl phosphine lithium base (2.40 g, 12.5 mmol) and (chloromethyl) dimethylchlorosilane (0.86 g, 6.0 mmol) in n-hexane (50 mL) was stirred for 3 h at −30°C using acidic ethanol. At room temperature, the solution was filtrated and evaporated. The residue was re-dissolved in n-hexane subsequently and filtered to remove insoluble material to afford a yellow oily substance. Upon recrystallization from n-hexane, yellowish crystalline product was obtained. Yield = 1.18 g (2.7 mmol, 45 %). 1H NMR (400 MHz, C6D6) δ(ppm) 7.53 (td, J = 7.8, 1.4 Hz, 4H), 7.41–7.29 (m, 4H), 7.10–6.90 (m, 12H), 1.55 (d, J = 2.3 Hz, 2H), 0.11 (d, J = 4.6 Hz, 6H). 31P NMR (162 MHz, C6D6) δ(ppm) −22.97 (d, J = 17.6 Hz), −55.86 (d, J = 17.5 Hz). 13C NMR (101 MHz, C6D6) δ(ppm) 143.18 (d, J = 16.1 Hz, 2C, ipso-Cquat PPh2), 138.05 (d, J = 16.9 Hz, 4C, ortho-CHPPh2), 136.19 (d, J = 17.3 Hz, 2C, ipso-Cquat PPh2), 134.73 (d, J = 19.8 Hz, 4C, ortho-CHPPh2), 130.48 (dd, J = 17.3, 6.7 Hz, 12C, meta/para-CHPPh2), 15.49 (dd, J = 33.5, 11.7 Hz, CH2), 0.14 (dd, J = 11.8, 4.8 Hz, 2C, CH3).
Synthesis of bis(diphenyl phosphine methyl) dimethyl silane(L3)
Bis(chloromethyl)dimethylsilane (0.79 g, 5.0 mmol) was reacted with diphenyl phosphine lithium base (1.94 g, 10.1 mmol) to give crystalline yellowish. Yield = 1.21 g (2.6 mmol, 52 %). 1H NMR (400 MHz, CDCl3) δ(ppm) 7.44–7.33 (m, 8H), 7.31–7.18 (m, 12H), 1.27 (s, 4H), −0.19 (s, 6H). 31P NMR (162 MHz, C6D6) δ −22.75(s), 13C NMR (101 MHz, CDCl3) δ 141.68 (d, J = 14.1 Hz, 4C, ipso-Cquat PPh2), 133.25 (d, J = 19.8 Hz, 8C, ortho-CHPPh2), 128.98 (dd, J = 11.2, 7.7 Hz, 12C, meta/para-CHPPh2), 15.11 (dd, J = 29.7, 4.7 Hz, 2C, CH2), 0.18 (dd, J = 40.2, 35.4 Hz, 2C, CH3).
Synthesis of metal complexes
All complexes were synthesized under a dry N2 atmosphere in oven-dried flasks using standard Schlenk techniques.
L1/NiBr2 complex
A dichloromethane solution (20 mL) of L1 (0.214 g, 0.5 mmol) was added to a dichloromethane solution (30 mL) of NiBr2DME (0.154 g, 0.5 mmol). The reaction mixture turned dark brown immediately and was allowed to stir for 24 h. The solvent was evaporated using a cold trap and vacuum pump and then the solid residue was washed three times with n-hexane. Drying in vacuo provided a dark brown powder. Yield = 0.162 g (50 %). Analytically calculated for C26H26Br2NiP2Si: C, 48.22; H, 4.02. Found: C, 48.26; H, 4.05. MALDI-TOF–MS: m/z = [M + 2H−2Br]+ = 489.1 (Scheme 1).

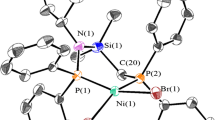
Structures of complex C1, C2 and C3
L2/NiBr2 complex
L2 (0.220 g, 0.5 mmol) and [NiBr2(DME)] (0.154 g, 0.5 mmol) were dissolved in CH2Cl2 (20 mL) and gained a brown solution. The mixture was allowed to stir for 24 h. After evaporation and purification, a brown powder was obtained. Yield: 0.262 g (79 %). Analytically calculated for C27H28Br2NiP2Si: C, 49.01; H, 4.24. Found: C, 49.06; H, 4.27. MALDI-TOF–MS: m/z = [M + Na-81Br]+ = 603.3.
L3/NiBr2 complex
L3 (0.228 g, 0.5 mmol) and [NiBr2(DME)] (0.154 g, 0.5 mmol) were dissolved in CH2Cl2 (20 mL) and gained a brown solution. The mixture was allowed to stir for 24 h. After evaporation and purification, a brown powder was obtained. Yield: 0.190 g (56 %). Analytically calculated for C28H30Br2NiP2Si: C, 49.77; H, 4.44. Found: C, 49.82; H, 4.48. MALDI-TOF–MS: m/z = [M−Br]+ = 595.1.
Molecular structure of C3
The molecular structure of complex 3 was determined by an X-ray diffraction study and the results were shown in Fig. 1. Crystallographic data and structural refinement parameters are given in Table 1. Selected bond lengths and bond angles of these complexes are listed in the captions for figures. The crystal structures of the complex confirm a two coordinated nickel metal center, which adopts an almost ideal planar geometry.s

The molecular structure of C3
General oligomerization procedure
The 140 mL Lab of Grest transparent glass reactor was heated in the high temperature drying oven for 2 h at 105 °C before use. Subsequently a magneton was placed inside the reactor, and then the reactor was heated in an oil bath to the temperature required for the oligomerization reaction. The reactor was equipped with a thermocouple, a pressure meter, and needle valves for injections. The reactor was vacuumed and then charged with high-purity N2, this process was repeated three times. Then repeat the above operation with ethylene. After that toluene solution of the catalyst precursor and the co-catalyst MAO were introduced into the reactor before the reactor was charged with 1.0 MPa of ethylene. The reaction was run for the required time (generally 30 min), after which the ethylene feed was turned off. Once the venting was completed, the reaction was quenched with acidic ethanol, and the product sampled for GC analyses. With n-heptane as internal standard,we calculate the catalytic activity and selectivity of the product in ethylene oligomerization (Table 2).
f is a mass correction factor; ms is the mass of n-heptane, g; mi is the mass of product extracted, g; Wt is the mass of the product, g; As is the peak area of n-heptane; Ai is the peak area of product extracted; t is the reaction time, h; n(Ni) is the moles of metal catalyst activity center, μmol.
M is the mass of α-olefin, g.
Results and discussion
Effect of reaction temperature on catalytic properties for the oligomerization of ethylene
Table 3 displays a steady increase in catalytic activity of C2 and C3 with the increase of temperature from 30 to 45 °C, which decreased significantly at temperature above 45 °C. As for C1, similar trend appeared at 75 °C. It indicates that the reaction temperature is an important parameter that affects the catalytic activity of C1, C2 and C3 complexes for ethylene oligomerization. It can be attributed to the lower ethylene solubility in toluene and higher deactivation rate of active site at high temperature [19]. The catalytic properties of C1, C2 and C3 are strongly affected by the ligand structure. C2 has higher catalytic activity toward ethylene dimerization than C1 and C3. There appears to be some general correlation between P–Ni–P bite angle. Complicated interaction between bridge structure (the number of carbon spacer ligands) and bridging atom is evident. The asymmetric structure of C2 may be more reasonable. The different activities obtained using diphosphines with C1, C2 and C3 suggest that ligand rigidity and/or electronic effects play an important role in catalyst performance as well. The 1-butylene selectivity of C3 is higher than that of C1 and C2 [16].
Effect of Al/Ni molar ratio on catalytic properties for the oligomerization of ethylene
The effect of Al/Ni molar ratios on catalytic properties is investigated and the results are listed in Table 4. The optimum Al/Ni molar ratio for C1, C2 and C3 were previously found to be 700:1. The activities of C1–C3 were 5.01, 31.2, and 2.98 × 105 g/(molNi h). All of the catalytic activity increased obviously when the Al/Ni molar ratio was added up to 700. The reason was that an optimal amount of MAO was needed to effectively activate the catalyst. At first, a part of MAO were consumed for eliminating residual oxygen and water in the reaction system when the less dosage of MAO and remaining amount is not enough to form catalytic active species with nickel and other catalyst components. The catalytic activity decreased subsequently with the molar ratio of Al/Ni further increasing. The reason could be that the excessive MAO has a great influence on the formation of active metal species, it might interfere with their formation or cause their over-reduction [20, 21]. The selectivity toward butylene did not change notably and remained relatively stable. C1 has slightly higher selectivity toward butylene than C2 and C3.
Effect of the reaction time on catalytic properties
Table 5 shows the catalytic activity and selectivity obtained at the different reaction times ranging from 15 to 60 min. The results indicate that the reaction time has a slight effect on the activity for the C1 and C3. C2 was different from other complexes, its activity was significantly affected by reaction time. The selectivity to C4 olefins is high and stable. The reaction time also influenced product distribution. By varying the reaction time from 15 to 60 min, chain isomerization transfer happened which was always relative to chain propagation. This can be seen from the data listed in Table 5.
By investigating the catalytic performance of complexes C1, C2, and C3, it was found out that the structure of the ligand has important effects on the activity and selectivity of the catalyst. Reaction parameters such as reaction time, temperature also resulted in different catalytic activities and selectivities.
Conclusion
We have synthesized novel ligands with variation of steric and electronic (L1, L2 and L3, respectively). Three novel nickel complexes (C1, C2 and C3) based on corresponding ligands have been prepared and evaluated for ethylene oligomerization under MAO activation. All nickel complexes catalyzed oligomerization of ethylene to butylene and hexene. Ligand structure with different length of carbon bridge has important influence on catalytic activity. With the increase of reaction temperature, catalytic activity and product selectivity were decreased. The excessive co-catalysts might interfere with the formation of active metal species or cause their over-reduction, which will decrease the catalyst activity. From the research, under the optimum conditions (45 °C, n(Al)/n(Ni) = 700, 30 min), Catalytic activity of C2 can be achieved 3.12 × 106 g mol−1 (cat.) h−1.
References
Malgas-Enus R, Mapolie SF (2014) Inorg Chim Acta 409:96–105
Boffa LS, Novak BM (2000) Chem Rev 100:1479–1494
Mecking S (2001) Angew Chem Int Ed 40:534–540
Gibson VC, Spitzmesser SK (2003) Chem Rev 103:283–316
Malinoski JM, Brookhart M (2003) Organometallics 22:5324–5335
Chen Q, Yu J, Huang J (2007) Organometallics 26:617–625
Ittel SD, Johnson LK, Brookhart M (2000) Chem Rev 100:1169–1204
Cornils B, Herrmann WA (1996) Applied homogeneous catalysis with organometallic compounds. VCH, Weinheim
Breuil P-AR, Magna L, Olivier-Bourbigou H (2015) Catal Lett 145:173–192
Boudier A, Breuil P-AR, Magna L, Olivier-Bourbigou H, Braunstein P (2012) J Organomet Chem 718:31–37
Cooley NA, Green SM, Wass DF, Heslop K, Orpen AG, Pringle PG (2001) Organometallics 20:4769–4771
Dennett JN, Gillon AL, Heslop K, Hyett DJ, Fleming JS, Lloyd-Jones CE, Orpen AG, Pringle PG, Wass DF, Scutt JN (2004) Organometallics 23:6077–6079
Boulens P, Lutz M, Jeanneau E, Olivier-Bourbigou H, Reek JN, Breuil PAR (2014) Eur J Inorg Chem 2014:3754–3762
Keim W (2013) Angew Chem Int Ed 52:12492–12496
Boulens P, Pellier E, Jeanneau E, Reek JN, Olivier-Bourbigou Hln, Breuil P-AR (2015) Organometallics 34:1139–1142
Overett MJ, Blann K, Bollmann A, de Villiers R, Dixon JT, Killian E, Maumela MC, Maumela H, McGuinness DS, Morgan DH (2008) J Mol Catal A 283:114–119
Sun Y, Chen Y, Mao G, Ning Y, Jiang T (2014) Chin Sci Bull 59:2613–2617
Jiang T, Chen H, Ning Y, Chen W (2006) Chin Sci Bull 51:521–523
Guo C-Y, Xu H, Zhang M, Zhang X, Yan F, Yuan G (2009) Catal Commun 10:1467–1471
Abbo HS, Titinchi SJ (2013) Molecules 18:4728–4738
Wang T, Dong B, Chen Y-H, Mao G-L, Jiang T (2015) J Organomet Chem 798:388–392
Acknowledgments
This study was supported by the National Natural Science Foundation of China (U1162114), Program for New Century Excellent Talents in University (NCET-07-0142) and the Provincial Key Laboratory of Oil & Gas Chemical Technology (HXHG2012-04).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Meng, X., Zhang, L., Chen, Y. et al. Silane-bridged diphosphine ligands for nickel-catalyzed ethylene oligomerization. Reac Kinet Mech Cat 119, 481–490 (2016). https://doi.org/10.1007/s11144-016-1056-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-016-1056-z