
Abstract
An iridium(III) complex [Ir(ppy)2(MHPIP)]PF6 (ppy = 2-phenylpyridine, MHPIP = 2-(1-methyl-1H-pyrazol-3-yl)-1H-imidazo[4,5-f][1, 10]phenanthroline, Ir-1) was synthesized and characterized by elemental analysis, IR, 1H NMR and 13C NMR. The in vitro cytotoxic activities of the free proligand MHPIP and the complex Ir-1 against HepG2, A549, BEL-7402, SGC-7901 and normal LO2 cells were evaluated by the MTT method. MHPIP has no cytotoxic activity toward the selected cell lines, while Ir-1 shows a moderate cytotoxic effect against HepG2. This complex also displays no cytotoxicity against normal LO2 cells, with an IC50 of more than 200 µM. The apoptosis of HepG2 cells induced by the complex was studied with AO/EB and DAPI staining methods, which showed that the complex can effectively induce apoptosis. A comet assay was performed by gel electrophoresis, and the results further show that the complex can cause apoptosis. The level of reactive oxygen species, mitochondrial membrane potential, autophagy, intracellular Ca2+ levels and cell invasion were investigated by fluorescence microscopy, and the cell cycle arrest was studied by flow cytometry. The expression of caspase and Bcl-2 family proteins was investigated by western blot. The results of these experiments indicate that Ir-1 accumulates preferentially in the mitochondria of HepG2 cells and induces apoptosis through inhibition of the PI3K/AKT/mTOR pathway.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma is usually fatal, while cellular carcinoma is the third leading cause of death in the world [1]. Therefore, the search for effective anticancer drugs remains a global research priority. Transition metal complexes are one such area of study, among which ruthenium complexes appear to be particularly interesting compounds. Presently, one ruthenium complex, namely KP1019 ([IndH][trans-RuCl4(Ind)2], where Ind = indazole), has entered clinical trials [2]. On the other hand, iridium-based compounds show a wide range of biological activities. Espinosa reported that iridium complexes can inhibit cell growth through DNA interactions [3]. Apoptosis, or type I programmed cell death, is characterized by cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation and chromosomal DNA fragmentation [4, 5]. Apoptosis plays a significant role in chemotherapies against all sorts of cancers [6]. The classical pathway of apoptosis can be subdivided into extrinsic and intrinsic components, which are triggered by death receptors or by various mitochondrial stimuli, respectively. On the other hand, autophagy, or type II programmed cell death, is an evolutionary conserved system that induces the degradation of cytoplasmic contents in a lysosome-dependent manner [7]. Autophagy plays important roles in many physiological and pathophysiological processes, such as suppressing apoptosis, innate immune response and subsequent recycling of cellular products [8]. Recent studies have indicated that many chemotherapeutics known to induce apoptosis also activate autophagy [9]. However, the interplay between apoptosis and autophagy is quite complex and sometimes contradictory in cancer development and treatment. In some cellular settings, autophagy can act as a protector, preventing cells from undergoing apoptosis by promoting cell survival, while in others, it can induce cell apoptosis [10, 11].
Reactive oxygen species (ROS) have toxic effects on various molecular targets including proteins, lipids, and DNA, leading to apoptosis and inflammatory response [12, 13]. Accumulating evidence suggests that ROS are important signaling molecules, which play a critical role in inducing both cell apoptosis and autophagy. Recent studies have also shown that some antitumor drugs can activate apoptosis and autophagy through inhibition of the PI3K/AKT/mTOR pathway [14]. In our previous work [15], we found that ruthenium(II) complexes of ligand MHPIP (MHPIP = 2-(1-methyl-1H-pyrazol-3-yl)-1H-imidazo[4,5-f] [1, 10] phenanthroline) induce apoptosis in HepG2 through a ROS-mediated mitochondrial dysfunction pathway. Substituted pyrazole ring derivatives exhibit a broad spectrum of biological activities including antimicrobial, herbicidal, antitumor and anti-inflammatory activities [16,17,18,19]. To obtain more insight into the anticancer activity and mechanism of metal complexes, in this paper, a new iridium(III) complex [Ir(ppy)2(MHPIP)]PF6 (ppy = 2-phenylpyridine, Ir-1, Scheme 1), containing a pyrazole ring, was synthesized and characterized by elemental analysis, IR, ESI–MS, 1H NMR and 13C NMR. The effects and potential mechanism of Ir-1-induced autophagy and apoptosis in HepG2 cells were investigated, and ROS-mediated PI3K/AKT/mTOR signaling pathways were identified in the autophagy and apoptosis.

The synthetic route of ligand and complex
Experimental
Materials and methods
All reagents and solvents were purchased commercially and used without further purification unless otherwise noted. Ultrapure Milli-Q water was used in all experiments. DMSO and RPMI 1640 were purchased from Sigma. 1,10-Phenanthroline was obtained from the Guangzhou Chemical Reagent Factory. HeLa (human cervical cancer), A549 (human lung carcinoma), HepG2 (human hepatocellular carcinoma), BEL-7402 (human hepatocellular carcinoma), SGC-7901 (human gastric adenocarcinoma), SiHa (human cervical carcinoma) and normal LO2 (human liver cell) cells were purchased from the American Type Culture Collection. IrCl3·3H2O was purchased from the Kunming Institution of Precious Metals. Microanalyses (C, H, and N) were obtained with a PerkinElmer 240Q elemental analyzer. Electrospray ionization mass spectra (ESI–MS) were recorded on an LCQ system (Finnigan MAT, USA) using acetonitrile as mobile phase. The spray voltage, tube lens offset, capillary voltage and capillary temperature were set at 4.50 kV, 30.00, 23.00 V and 200 °C, respectively, and the quoted m/z values are for the major peaks in the isotope distribution. 1H and 13C NMR spectra were recorded on a Varian-500 spectrometer with DMSO-d6 as solvent and tetramethylsilane (TMS) as an internal standard at 500 MHz at room temperature.
Synthesis of complex Ir-1
A mixture of cis-[Ir(ppy)2Cl]2 [20] (0.16 g, 0.15 mmol) and MHPIP (0.09 g, 0.3 mmol) [15] in a mixture of dichloromethane plus methanol (2:1 v/v; 42 mL total) was refluxed under argon for 6 h to give a clear yellow solution. After cooling, a yellow precipitate was obtained by dropwise addition of saturated aqueous NH4PF6 with stirring at room temperature over 2 h. The crude product was purified by column chromatography on neutral alumina with a mixture of CH2Cl2–acetone (1:1, v/v) as eluent. The solvent was removed under reduced pressure to give a yellow powder. Yield: 74%. Anal. Calc for C39H28N8IrPF6: C, 49.52, H, 2.98, N, 11.85%. Found: C, 49.63, H, 2.89, N, 11.77%. IR (KBr, cm−1): 3374s, 3044w, 2964s, 1608s, 1583w, 1562m, 1478s, 1438s, 1417s, 1365w, 1306m, 1163m, 1078w, 1013w, 845s, 758s, 558s. 1H NMR (DMSO-d6): 9.10 (d, 2H, J = 8.0 Hz), 8.29 (s, 1H), 8.10 (dd, 3H, J = 5.0, J = 5.0 Hz), 7.90 (d, 2H, J = 8.0 Hz), 7.72–7.65 (m, 6H), 7.35 (d, 2H, J = 5.5 Hz), 7.24 (s, 1H), 7.05 (t, 2H, J = 7.5 Hz), 6.94 (t, 2H, J = 6.5 Hz), 6.84 (t, 2H, J = 6.5 Hz), 6.38 (d, 2H, J = 7.0 Hz), 3.83 (s, 3H). 13C NMR (DMSO-d6, 125 MHz): 168.20, 150.49, 149.84, 148.75, 148.19, 144.18, 143.81, 138.62, 138.21, 132.87, 132.07, 131.08, 130.48, 126.26, 125.03, 124.61, 123.36, 122.93, 119.80, 113.68, 39.42. ESI–MS (CH3CN): m/z 802.1 [M-PF6]+.
Cell viability assay
Inhibition of cell proliferation by [Ir(ppy)2(MHPIP)]PF6 (Ir-1) was measured by 3-(4,5-dimethylthiazole-2-yl)-2,5-biphenyl tetrazolium bromide (MTT) assay. Cells were placed in 96-well microassay culture plates (8 × 103 cells per well) and grown overnight at 37 °C in a 5% CO2 incubator. The test compounds were then added to the wells to achieve final concentrations ranging from 10−6 to 10−4 µM. Control wells were prepared by addition of culture medium (100 µL). The plates were incubated at 37 °C in a 5% CO2 incubator for 48 h. Upon completion of the incubation, stock MTT dye solution (20 µL, 5 mg mL−1) was added to each well. After 4 h, buffer (100 µL) containing dimethylformamide (50%) and sodium dodecyl sulfate (20%) was added to solubilize the MTT formazan. The optical density of each well was measured with a microplate spectrophotometer at a wavelength of 490 nm. The IC50 values were determined by plotting the percentage of cell viability versus concentration on a logarithmic graph and reading off the concentration at which 50% of cells remained viable relative to the control. Each experiment was repeated at least three times to obtain mean values.
Apoptosis studies with AO/EB and DAPI staining
HepG2 cells (2 × 105) were exposed to 25.0 μM of the complex and cultured in RPMI (Roswell Park Memorial Institute) 1640 with 10% of fetal bovine serum (FBS) and incubated at 37 °C in 5% CO2 for 24 h. The cells were washed with ice-cold phosphate buffer saline (PBS) and fixed with formalin (4%, w/v). Cell nuclei were counterstained with acridine orange (AO) and ethidium bromide (EB) (AO: 100 μg/mL, EB: 100 μg/mL) or DAPI (100 μg/mL) for 10 min. The cells were imaged with a fluorescence microscope (Nikon, Yokohama, Japan) with excitation at 350 nm and emission at 460 nm.
Comet assay
Comet assays were performed according to the literature [21]. To access DNA damage, HepG2 cells in culture medium were incubated with 25 or 50 µM of the complex for 24 h at 37 °C. Control cells were also incubated in the same time. The cells were harvested by trypsinization at 24 h. A total of 100 mL of 0.5% normal agarose in PBS was dropped gently onto a fully frosted microslide, covered immediately with a coverslip, and then kept at 4 °C for 10 min. The coverslip was removed after the gel had set. A 50 mL portion of the cell suspension (200 cells per mL) was mixed with 50 mL of 1% low-melting agarose preserved at 37 °C. A total of 100 mL of this mixture was applied quickly on top of the gel, coated over the microslide, covered immediately with a coverslip, and then kept at 4 °C for 10 min. The coverslip was again removed after the gel had set. A third coating of 50 mL of 0.5% low-melting agarose was placed on the gel and allowed to set at 4 °C for 15 min. After solidification of the agarose, the coverslips were removed, and the slides were immersed in an ice-cold lysis solution (2.5 mM NaCl, 100 mM EDTA, 10 mM Tris, 90 mM sodium sarcosinate, NaOH, pH 10, 1% Triton X-100 and 10% DMSO) and kept in a refrigerator at 4 °C for 2 h. All of the above operations were performed under low lighting conditions to avoid additional DNA damage. The slides, after removal from the lysis solution, were placed horizontally in an electrophoresis chamber. The reservoirs were filled with an electrophoresis buffer (300 mM NaOH, 1.2 mM EDTA) until the slides were just immersed in it, and the DNA was allowed to unwind for 30 min. Electrophoresis was then carried out at 25 V and 300 mA for 20 min. After electrophoresis, the slides were removed and washed thrice in a neutralization buffer (400 mM Tris–HCl, pH 7.5). Cells were stained with 20 mL of EB (20 mg mL−1) in the dark for 20 min. The slides were washed in chilled distilled water for 10 min to neutralize the excess alkali, air-dried and scored for comets by fluorescence microscopy.
ROS detection
HepG2 cells were seeded into six-well plates (Costar, Corning Corp, New York, USA) at a density of 2 × 105 cells per well and incubated for 24 h. The cells were cultured in RPMI 1640 supplemented with 10% FBS and incubated at 37 °C in 5% CO2. The medium was removed and replaced with fresh medium (final DMSO concentration, 0.05% v/v) containing different concentrations of the complex; after 24 h, the cells were stained with 20 µM DCFH-DA in PBS for 30 min in the dark. Finally, the cells were washed twice with PBS, and then the cells were imaged under a fluorescent microscope.
Location of the complex in the mitochondria
HepG2 cells were placed in 24-well microassay culture plates (4 × 104 cells per well) and grown overnight at 37 °C in a 5% CO2 incubator. 25.0 µM of the complex was added, and the cells were kept at 37 °C in a 5% CO2 incubator for 4 h and further co-incubated with MitoTracker® Deep Red FM (100 nM) at 37 °C for 1 h. Upon completion of the incubation, the wells were washed three times with ice-cold PBS. After discarding the culture medium, the cells were imaged under a fluorescence microscope.
Mitochondrial membrane potential assay
HepG2 cells were treated for 24 h with different concentrations of the complex in 12-well plates and then washed three times with cold PBS. The cells were detached with trypsin–EDTA solution. Collected cells were incubated for 20 min with 1 µg/mL of JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanineiodide) in culture medium at 37 °C in the dark and then immediately centrifuged to remove the supernatant. Cell pellets were suspended in PBS and imaged under a fluorescence microscope. The ratio of green/red fluorescence intensity was determined by flow cytometry.
Matrigel invasion assay
A BD Matrigel invasion chamber was used to investigate the cell invasion according to the manufacturer’s instructions. HepG2 cells (4 × 104) in serum-free media with different concentrations of the complex were seeded in the top chamber of the two-chamber Matrigel system. RPMI-1640 (20% FBS) was added as a chemo-attractant into the lower chamber. Cells were allowed to invade for 24 h. After incubation, non-invading cells were removed from the upper surface and cells on the lower surface were fixed with 4% paraformaldehyde and stained with 0.1% of crystal violet. The membranes were photographed, and the invading cells were counted under a light microscope. The mean values from three independent assays were calculated.
Measurement of intracellular Ca2+ level
HepG2 cells were treated with different concentrations of Ir-1 for 4 or 6 h, and then the cells were stained with Fluo-3 AM for 30 min at 37 °C in the dark, washed with PBS three times and then incubated for an additional 20 min with PBS at 37 °C to ensure that Fluo-3 AM had been completely transformed into Fluo-3, which can specifically bind to Ca2+ and has a strong fluorescence with an excitation wavelength of 488 nm. The cell nuclei were stained with DAPI at 37 °C. Finally, an ImageXpress Micro XLS system was used to observe fluorescence, and a Multi-Wavelength Cell Scoring module was used to analyze the data. The integrated intensity/cell was used to measure the levels of Ca2+. The fluorescence intensity of each cell was calculated as the total fluorescence intensity divided by the number of cells.
Cell cycle arrest by flow cytometry
HepG2 cells were seeded into six-well plates (Costar, Corning Corp, New York, USA) at a density of 2 × 105 cells per well and incubated for 24 h. The cells were cultured in RPMI 1640 supplemented with 10% of FBS and incubated at 37 °C in 5% CO2. The medium was removed and then replaced with fresh medium (final DMSO concentration, 0.05% v/v) containing different concentrations of the complex. After incubation for 24 h, the cell layer was trypsinized, washed with cold PBS and fixed with 70% ethanol. 20 µL of RNAse (0.2 mg/mL) and 20 µL of propidium iodide (0.02 mg/mL) were added to the cell suspensions, which were then incubated at 37 °C for 30 min. The samples were analyzed with an FACSCalibur flow cytometer. The number of cells analyzed for each sample was 10,000 [22].
Induction of autophagy
HepG2 cells were incubated with the fluorescent probe MDC to quantify the induction of autophagy [23]. HepG2 cells were seeded onto chamber slides in 12-well plates and incubated for 24 h. The cells were cultured in RPMI 1640 supplemented with 10% FBS and incubated at 37 °C in 5% CO2. The medium was removed and replaced with fresh medium (final DMSO concentration, 0.05% v/v) containing different concentrations of the complex and then incubated for 24 h. The medium was removed again, and the cells were washed twice with ice-cold PBS. The cells were stained with MDC (monodansylcadaverine) solution (50 µM) for 10 min and then washed twice with PBS. The cells were observed and imaged under a fluorescence microscope. The effect of the complexes on the expression of Beclin-1 and LC3 proteins was assayed by western blot.
Effect of autophagy and NAC on cell viability
HepG2 cell viability was evaluated using the MTT method. Cells were placed in 96-well microassay culture plates (8 × 104 cells per well) and cultured overnight at 37 °C in a 5% CO2 incubator. The cells were pretreated with or without 3-methyladenine (3-MA, 1 mM) or N-acetyl-l-cysteine (NAC, 10 mM) for 2 h, followed by different concentrations of Ir-1 for 24 h. After incubation, cells were incubated with MTT (0.5 mg/ml) for 4 h at 37 °C. Upon completion of the incubation, 100 μL DMSO was added to solubilize the MTT formazan. The optical density of each well was then measured with a microplate spectrophotometer at a wavelength of 490 nm. The viability (%) of cell growth was calculated by the formula:
where Atreatment group is the mean OD value of cells treated with the iridium complex (6.25 µM → 100 µM) and Acontrol is the mean OD value of untreated cells. Each experiment was repeated three times to obtain mean values.
Western blot analysis
HepG2 cells (2 × 105) were treated with different concentrations of the complex for 24 h. The cells were harvested in lysis buffer and centrifuged at 13,000g for 20 min. The protein concentration of the supernatant was determined by BCA (bicinchoninic acid) assay. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis was done by loading equal amounts of proteins per lane. Gels were then transferred to poly(vinylidene difluoride) membranes (Millipore) and blocked with 5% nonfat milk in TBST (20 mM Tris–HCl, 150 mM NaCl, 0.05% Tween 20, pH 8.0) buffer for 1 h. The membranes were then incubated with primary antibodies at 1:5000 dilution in 5% nonfat milk overnight at 4 °C and washed four times with TBST for a total of 30 min. After this, the secondary antibodies were conjugated with horseradish peroxidase at 1:5000 dilution for 1 h at room temperature and then washed four times with TBST. The blots were visualized with the Amersham ECL Plus western blotting detection reagents according to the manufacturer’s instructions.
Data analysis
All data are expressed as mean ± SD. Statistical significance was evaluated by a t test. Differences were considered to be significant when a *P value was < 0.05.
Results and discussion
Synthesis and characterization
The proligand MHPIP was prepared according to the literature [20]. The complex [Ir(ppy)2(MHPIP)]PF6 (Ir-1) was synthesized by the reaction of cis-[Ir(ppy)2Cl]2 with MHPIP in a mixture of dichloromethane and methanol and purified by column chromatography. The UV–Vis and luminescence spectra of the complex in PBS solution are shown in Fig. 1. The maximum absorbance of Ir-1 is observed at 289 nm; it is also luminescent in PBS solution at ambient temperature, with a maximum emission at 564 nm. In the IR spectrum, a peak at 2964 cm−1 is assigned to the C=C stretching vibrations. Peaks at 3.83 ppm in the 1H NMR and 39.42 ppm in the 13C NMR spectra are attributed to the hydrogen and carbon atoms of the methyl group, respectively. In the ESI–MS spectrum, a peak at a m/z value of 802.1 corresponds to the ion peak of [M-PF6]+.

UV–Vis (a) and luminescence (b) spectra of the complex in PBS solution at room temperature
Cytotoxicity of Ir-1
The in vitro cytotoxicity of a test compound is an important consideration in anticancer drug studies. The cytotoxicity of free MHPIP and Ir-1 against SGC-7901, HeLa, BEL-7402, HepG2, A549 and normal LO2 cell was therefore assayed using the MTT method. The cells were treated with various concentrations of MHPIP or Ir-1 for 48 h, giving the IC50 values listed in Table 1. As expected, free MHPIP displays no cytotoxic activity toward the selected cell lines, whereas Ir-1 shows different cytotoxic activities toward different cancer cell lines. Ir-1 complex is toxic to HepG2 cells, with an IC50 value of 39.5 ± 6.0 µM, but has no cytotoxic activity against the other cancer cells. Thus, we consider that the complex is selective for HepG2 cancer cells. Additionally, the complex only kills cancer HepG2 cells, but has low cytotoxicity toward normal LO2 cells. This result is rarely observed because most metal complexes are toxic toward both cancer cells and normal cells. In light of these results, the HepG2 cell line was selected for the following experiments.
Ir-1 induces apoptosis in HepG2 cells
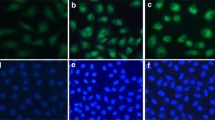
Induction of tumor cell apoptosis is a very important way for many anticancer drugs to exert their function [24]. Phosphatidyl serine (PS) is located in the plasma membrane of the closed beta in normal cells; in apoptotic cells, PS is transferred to the outside of the plasma membrane. Apoptosis in HepG2 cells was studied using the acridine orange (AO)/ethidium bromide (EB) staining method. It is well known that AO can pass through the cell membrane, but EB cannot. As shown in Fig. 2A, in the control experiment (a), living cells were stained bright green. Upon exposure of HepG2 cells to 25 (b) or 50 µM (c) of Ir-1 for 24 h, green apoptotic cells containing apoptotic features were observed. In addition, the apoptosis was also assayed using DAPI (d–f) and Hoechst 33,258 (g–i) staining methods. After the treatment of HepG2 cells (d, g) with 25 (e, h) or 50 µM (f, i) of Ir-1 for 24 h, the cell nuclei were stained bright blue. Annexin V, a Ca2+-dependent protein with high affinity for PS, has been used to identify apoptosis at an early stage [25]. To quantitatively determine the effect of the concentration of the complex on apoptosis, the apoptosis was investigated using FACS analysis of Annexin V-FITC/PI double staining. As shown in Fig. 2B, when HepG2 cells (a) were incubated with 25 (b) or 50 (c) µM of Ir-1 for 24 h, the proportions of apoptotic cells were 3.76 and 4.00%, respectively. Hence complex Ir-1 induces weak apoptosis in a concentration-dependent manner.

A Apoptosis in HepG2 cells (a, d) exposure to 25 (b, e) and 50 (c, f) µM of complex for 24 h and the cells were stained with AO/EB and DAPI, respectively. B The percentage of apoptotic cell was determined by flow cytometry. HepG2 cells (a) exposure to 25 (b) and 50 (c) µM of Ir-1 for 24 h
DNA damage studies
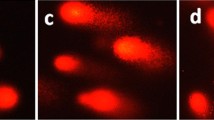
The ability of the complex to induce apoptosis was evaluated in HepG2 cells using the comet assay, which is used to study DNA fragmentation or damage [26]. The amount of DNA that migrates away from the nucleus is used to assess the extent of DNA damage. When a cell with damaged DNA is subjected to electrophoresis and then stained with EB, it appears as comet shaped [27]. As shown in Fig. 3, in the control experiment (a), no comet-like appearance was observed. On the contrary, the tail length and the amount of tailing DNA were significantly increased for both 25 and 50 μM of Ir-1 in a dose-dependent manner. The length of the comet tail represents the extent of DNA damage, which is regarded as a feature of apoptosis [27]. The results clearly indicate that the complex can induce DNA fragmentation, providing further evidence of apoptosis.

Comet assay of HepG2 cell (a) exposure to 25 (b) and 50 (c) µM of Ir-1 for 24 h
The cell invasion assay
Cell invasion, which is a critical component of the metastatic process and a major clinical challenge in cancer treatment, is responsible for the majority of cancer treatment failure and patient death [28]. To determine the effects of Ir-1 on the inhibition of HepG2 cell invasion, a Matrigel invasion assay was carried out. As shown in Fig. 4A, on treatment of HepG2 cells (a) with 25 (b) or 50 (c) µM of Ir-1 for 24 h, a significant decrease in the number of invasive cells was observed compared with the control. Use of 25 and 50 µM of Ir-1 inhibited cell invasion by 56.7% and 89.1%, respectively (Fig. 4B). Therefore, Ir-1 shows a concentration-dependent inhibition of cell invasion in HepG2 cells.

A Microscope images of invading HepG2 cells that have migrated through the Matrigel: the extent of inhibition of cell invasion by 25 (b) and 50 (c) µM complex against HepG2 (a) for 24 h. B The percentage of invading HepG2 cells induced by 25 and 50 µM of the complex for 24 h. *P < 0.05 represents significant differences compared with control
Location and changes in mitochondrial membrane potential
Mitochondrial membrane potential changes are one of the early events leading to functional alterations and are often associated with apoptosis [29, 30]. We have therefore investigated the localization of Ir-1 in the mitochondria using MitoTracker® Deep Red FM (Thermo Fisher, 100 nM) as red fluorescent probe. As shown in Fig. 5A, in the control experiment (a), the mitochondria were stained red. After the treatment of HepG2 cells with 25 µM of Ir-1 for 24 h, the complex emits green fluorescence (b). The overlay (c) of the red and the green images indicates that the complex interacts with the mitochondria. To study the effects of Ir-1 on the mitochondrial membrane potential, the change of potential ΔΨ (MMP) was detected using JC-1 as fluorescent probe. JC-1 forms aggregates that emit green fluorescence at low membrane potential and monomers that emit red fluorescence at high membrane potential [31]. As shown in Fig. 5B, in the control experiment (a), JC-1 emits red fluorescence. HepG2 cells were exposed to cccp (carbonyl cyanide-m-chlorophenyl hydrazone, b, positive control), 25 (c) or 50 (d) µM of Ir-1 for 24 h, and JC-1 emits bright green fluorescence. To quantitatively evaluate the effect of the concentration of Ir-1, the ratio of green/red fluorescence intensity was determined by flow cytometry. In the control (Fig. 5C), the ratio of green/red fluorescence was 1.8. Treatment of HepG2 cells with 25 or 50 µM of Ir-1 led to an increase in the ratio to 2.5 and 3.9, respectively. Hence, in the presence of Ir-1, the red fluorescent intensity decreases and green intensity increases, indicating that Ir-1 induces a decrease in the mitochondrial membrane potential in a concentration-dependent manner.

A The location of the complex in the mitochondria in HepG2 cell exposure to 25 µM of complex for 24 h. B The changes of mitochondrial membrane potential were studied after HepG2 (a) cells were treated with cccp (b, positive control) and 25 (c), 50 (d) µM of Ir-1 for 24 h and the cells were imaged under a fluorescent microscope. C The ratio of the green/red fluorescent intensity was determined after HepG2 cells were treated with 25 and 50 µM of the complex for 24 h. *P < 0.05 represents significant differences compared with control
Cell cycle arrest by flow cytometry
According to the content of DNA, the cell cycle can be divided into different parts: DNA synthesis (S), gap2/mitosis (G2/M), gap1 (G0/G1) and sub-G1 (including dead cells and cellular debris). The apoptosis of cancer cells is often associated with genomic DNA damage and cell cycle disturbance [27, 28]. The distribution of HepG2 cells crossing the different phases in the cell cycle was assayed by flow cytometry analysis. As shown in Fig. 6, in the control, the proportion of cells at G0/G1 phase is 43.73%, and upon exposure of the HepG2 cells to 25.0 or 50 μM of Ir-1, the proportion of cells at G0/G1 phase was 47.73% and 49.00%, respectively, accompanied by corresponding decreases in the S phase cells. Hence, the complex induces cell cycle arrest at the G0/G1 phase in HepG2 cells, in a concentration-dependent manner.

The cell cycle arrest in HepG2 cells exposed to 25 and 50 µM of Ir-1 for 24 h
Detection of intracellular Ca2+ levels
Increase in intracellular free Ca2+ is one of the primary triggers leading to the release of proapoptotic proteins from the mitochondria, ultimately leading to cell apoptosis [32,33,34]. The intracellular Ca2+ level was detected using Fluo-3 AM as a fluorescent probe, which can cross the cell membrane and be converted into Fluo-3 by intracellular esterases. The Fluo-3 binds specifically to Ca2+ and has a strong fluorescence. As shown in Fig. 7A, in the control experiment (a), no obvious fluorescent points are observed. When HepG2 cells were exposed to 25 (b) or 50 µM (c) of the complex for 4 h, a number of green fluorescent points were found, revealing that the complex can increase intracellular Ca2+ levels. Figure 7B shows that the fluorescence intensity increases with both increasing concentration and increasing exposure time of the cells to Ir-1.

A Intracellular Ca2+ levels were assayed after HepG2 cells were exposed to 25 and 50 µM of Ir-1 for 24 h. B The integrated fluorescent intensity/cell was determined after HepG2 cells were treated with different concentration of Ir-1 for 24 h. *P < 0.05 represents significant differences compared with control
Ir-1 induces autophagy in HepG2 cells
Autophagy is a relatively conserved metabolic pathway in cells. It is mainly used to maintain the stability of the intracellular environment [35], and autophagy is a key mechanism in various physiopathological processes including cell death and survival [36]. Recently, autophagy has become a major area of study in the treatment of various cancers [37, 38]. To examine whether or not Ir-1 can induce autophagy in HepG2 cells, we used MDC as a specific, in vivo fluorescent probe for autophagic vacuoles [39]. As shown in Fig. 8A, with increasing concentrations of the complex, the fluorescence intensity of the acidic autophagic vacuoles increases. To quantify this observation, MDC fluorescence intensity was determined by flow cytometry (Fig. 8B). In the control experiment (a), the fluorescence intensity of MDC is 2.92. When HepG2 cells were incubated with 25 µM (b) or 50 µM (c) of Ir-1 for 24 h, the MDC fluorescence intensity increased by 20.1 and 97.6 times compared to the control, respectively. Hence, the complex can induce autophagy in HepG2 cells in a concentration-dependent manner.

A Autophagy in HepG2 cell (a) was treated with 25 (b) and 50 (c) µM of Ir-1 for 24 h and the cells were stained with MDC. B MDC fluorescent intensity in the autophagy was determined by flow cytometry, while HepG2 cells (a) were exposed to 25 and 50 µM of Ir-1 for 24 h. C The conversion of LC3-I to LC3-II and expression of Beclin-1 protein in the absence or presence of 3-MA (D) or NAC (E) were assayed by western blot after HepG2 cells were incubated with 25 µM of Ir-1 for 24 h
We further examined the effects of Ir-1 on the expression of autophagy-associated proteins. Beclin-1 and LC3 are the central control proteins involved in the autophagic flux [40]. Beclin-1 acts in cooperation with class III-type phosphoinositide 3-kinase (class III PI3K, also known as Vps34) to regulate autophagosome formation and cargo recruitment [41]. LC3 is a structural protein of the autophagosomal membrane, and levels of LC3-B have been correlated with the extent of autophagosome formation [42]. As seen in Fig. 8C, when HepG2 cells were treated with Ir-1, the level of autophagosome (LC3-II) in the cytoplasm increased compared with the control. Moreover, the expression of Beclin-1 and the conversion of LC3-I to LC3-II were upregulated. The autophagy was also investigated in the presence of 3-MA or NAC (3-MA is an inhibitor to inhibit autophagy, and NAC is an inhibitor to inhibit the product of ROS). As shown in Fig. 8D, E, in the presence of 3-MA or NAC, the autophagy was inhibited, suggesting that autophagy is closely correlated with ROS levels. These findings show that Ir-1 can effectively induce autophagy in HepG2 cells.
Relation between autophagy and cell viability
To evaluate whether Ir-1 prevents or induces cell death by its effect on autophagy, HepG2 cells were treated with different concentrations of Ir-1 in the absence or presence of 3-MA or NAC for 24 h. As shown in Fig. 9A, in the presence of 3-MA, which is an inhibitor of autophagy, the cell viability decreased compared to treatment with Ir-1 alone. However, in the presence of NAC, which is an inhibitor of ROS production, the cell viability increased. To further investigate the relation between autophagy, ROS and apoptosis, HepG2 cells were exposed to 25 µM of Ir-1 for 24 h in the absence or presence of NAC or 3-MA. As shown in Fig. 9B, in the absence of NAC or 3-MA, the proportion of apoptotic cells is 4.00%. In the presence of NAC or 3-MA, the proportions of apoptotic cells are 3.08% and 7.18%, respectively. Therefore, ROS increases apoptosis and autophagy inhibits apoptosis in the presence of Ir-1.

A Cell viability was assayed in the presence of 3-MA or NAC by MTT method. B The apoptotic percentage in the cells was determined by flow cytometry after HepG2 cells were exposed to 50 µM of Ir-1 for 24 h
ROS-mediated Ir-induced apoptosis and autophagy
ROS play a critical role in regulation of cellular programs and signal transduction [43, 44].To investigate whether Ir-1 can increase the generation of ROS in HepG2 cells, intracellular ROS levels were assayed using DCFH-DA as a fluorescent probe. DCFH-DA is a cell permeating dye and is cleaved by intracellular esterases into its non-fluorescent form DCHF. DCHF is in turn oxidized by intracellular free radicals to produce a fluorescent product DCF. As shown in Fig. 10, in the control experiment (a), the DCF fluorescence intensity is 17.0. After treatment of HepG2 cells with 25.0 (c) or 50 µM (e) of Ir-1 for 24 h, the DCF fluorescence increased to 2.3 and 18.6 times that of the original, indicating that Ir-1 can increase intracellular ROS levels. To further investigate the relation between ROS and autophagy, HepG2 cells were exposed to 3-MA (b, another control), 25.0 µM Ir-1 + 3-MA (d) or 50.0 µM Ir-1 + 3-MA (f) for 24 h, whereupon the DCF fluorescence intensity increased by 28.5 and 29.1 times that of the control (HepG2 + 3-MA). This shows that autophagy inhibits the production of ROS. Ruthenium complexes of the type [Ru(η6-biphenyl)(azpy)I]+ (azpy = 2-(phenyldiazeny)pyridine) are relatively inert toward ligand substitution and appear to kill tumor cells by ligand-centered redox-mediated mechanisms [45]. Salder et al. reported that Os(II) arene complexes induced a dramatic increase in the levels of ROS in A549 lung cancer cells, suggesting that they interfere with the redox signaling pathways in cancer cells [46, 47]. In our previous work, we found that Ru(II) polypyridyl complexes induce apoptosis through an ROS-mediated mitochondrial dysfunction pathway [48,49,50]. These results show that [Ir(ppy)2(MHPIP)]+ also induces apoptosis through an ROS-mediated mitochondrial dysfunction pathway similar to those of Ru(II) polypyridyl complexes.

Intracellular ROS levels in HepG2 cells exposed to 25 and 50 µM of Ir-1 in the presence of 3-MA for 24 h and the DCF fluorescent intensity was determined by flow cytometry
Expression of caspase 3 and Bcl-2 family proteins
PARP cleavage is considered to be a hallmark of apoptosis [51]. Caspases are known to mediate the apoptotic pathway. The expression of PARP, caspase 3 and Bcl-2, Bad and Bax was therefore studied by western blot analysis. As shown in Fig. 11A, when HepG2 cells were incubated with 25.0 or 50.0 µM of Ir-1 for 24 h, PARP cleavage was observed. This indicates that the complex can induce apoptosis in HepG2 cells and also up-regulate the expression of caspase 3, Bad and Bax and down-regulate the expression of Bcl-2. The expression of these proteins was also investigated in the presence of 3-MA or NAC. As shown in Fig. 11B, C, 3-MA increases the expression of caspase 3, Bad and Bax, whereas NAC inhibits the expression of caspase 3, Bad and Bax in the presence of Ir-1. Thus, we consider that autophagy down-regulates the expression of caspase 3, Bad and Bax, while ROS have the opposite effect.

Western blot analysis of PARP, caspase 3, Bcl-2, Bad and Bax in HepG2 cells treated with 25 μM of Ir-1 in the absence a and presence of 3-MA b or NAC c for 24 h. β-actin was used as internal control
Apoptotic mechanism studies
The PI3K/AKT/mTOR signaling pathway is central to promote cell growth, motility, protein synthesis, survival and metabolism in response to hormones, growth factors and nutrients [52,53,54]. PI3K activates the serine/threonine kinase AKT, which in turn through a cascade of regulators results in the phosphorylation and activation of the serine/threonine kinase mTOR [53]. We used immunoblot to analyze the effect of Ir-1 on the PI3K/Akt/mTOR signaling pathway. As shown in Fig. 12A, upon exposure of HepG2 cells to the complex for 24 h, the levels of PI3K, pAKT and mTOR were significantly decreased in a dose-dependent manner. These data suggest that Ir-1 mediates the apoptosis and autophagy through inhibition of the PI3K/AKT/mTOR signaling pathway. Additionally, the effects of 3-MA or NAC on the expression of PI3K, AKT, pAKT and mTOR were also investigated. As shown in Fig. 12B, C, in the presence of 3-MA or NAC, the expression of PI3K, pAKT and mTOR was upregulated compared with that for Ir-1 alone. Hence, autophagy and ROS down-regulate the expression of PI3K, pAKT and mTOR. We conclude that Ir-1 induces apoptosis in HepG2 cells through inhibition of the PI3K/AKT/mTOR pathway.

PI3K/AKT/mTOR signal pathway assay in the absence or presence of 3-MA or NAC after 25 µM of Ir-1-treated HepG2 cells for 24 h
Conclusions
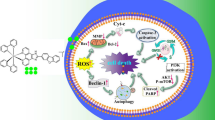
A new iridium(III) complex was synthesized and characterized. The complex displays moderate cytotoxic activity against HepG2 cells, inducing apoptosis. The comet assays show that the complex causes DNA damage. Additionally, the complex increases intracellular ROS levels, decreases the mitochondrial membrane potential and causes autophagy. It effectively inhibits cell invasion and growth at the G0/G1 phase. Overall, we consider that the complex induces apoptosis and autophagy through DNA damage, ROS-mediated mitochondria dysfunction, and inhibition of PI3K/AKT/mTOR pathways (Fig. 13). This work will be helpful for the further design and synthesis of new iridium(III) complexes as potent anticancer reagents.

The molecular mechanism of the complexes induced apoptosis in HepG2 cell
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) CA Cancer J Clin 61:69–90
Meng XJ, Leyva ML, Jenny M, Gross I, Benosman S, Fricker B, Harlepp S, Hébraud P, Boos A, Wlosik P, Bischoff P, Sirlin C, Pfeffer M, Loeffler JP, Gaiddon C (2009) Cancer Res 69:5458–5466
Ruiz J, Rodríguez V, Cutillas N, Samper KG, Capdevila M, Palacios Ò, Espinosa A (2012) Dalton Trans 41:12847–12856
Nagasaka A, Kawane K, Yoshida H, Nagata S (2010) Cell Death Differ 17:931–941
Burgess DJ (2013) Nat Rev Cancer 13:79
Zimmermann KC, Bonzon C, Green DR (2001) Pharmacol Ther 92:57–70
Klionsky DJ, Emr SD (2000) Science 290:1717–1721
Zhang Y, Yang JW, Ren X, Yang JM (2011) Autophagy 7:1557–1558
Li Y, Zhu H, Zeng X, Fan J, Qian X, Wang S, Wang Z, Sun Y, Wang X, Wang W, Ju D (2013) Mol Neurobiol 47:1000–1010
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A (2009) Cell Death Differ 16:966–975
Song X, Kim SY, Zhang L, Tang D, Bartlett DL, Kwon YT, Lee YJ (2014) Cell Death Dis 5:e1504
Zhang M, Harashima N, Moritani T, Huang W, Harada M (2015) PLoS ONE 10:e0127386
Qin G, Wu L, Liu H, Pang Y, Zhao C, Wu S, Wang X, Chen T (2015) Exp Cell Res 336:308–317
Li YC, He SM, He ZX, Li M, Yang Y, Pang JX (2014) Cancer Lett 344:239–259
Wan D, Tang B, Wang YJ, Guo BH, Yin H, Yi QY, Liu YJ (2017) Eur J Med Chem 139:180–189
Chen H, Li Z, Han Y (2000) J Agric Food Chem 48:5312–5315
Meazza G, Bettarini F, Porta LP, Piccardi P, Signorini E, Portoso D, Fornara L (2004) Pest Manag Sci 60:1178–1188
Park H, Lee K, Park S, Ahn B, Lee J, Cho HY, Lee K (2005) Bioorg Med Chem Lett 15:3307–3312
Bekhit AA, Ashour HMA, Guemei AA (2005) Arch Pharm 2338:167–174
Sprouse S, King KA, Spellane PJ, Watts RJ (1984) J Am Chem Soc 106:6647–6653
Lai SH, Jiang GB, Yao JH, Li W, Han BJ, Zhang C, Zeng CC, Liu YJ (2015) J Inorg Biochem 152:1–9
Lo KK, Lee TK, Lau JS, Poon WL, Cheng SH (2008) Inorg Chem 47:200–208
Contento AL, Xiong Y, Bassham DC (2005) Plant J 42:598–608
Chen TF, Wong YS (2008) Cell Mol Life Sci 65:2763–2775
Bai F, Ni B, Liu M, Feng Z, Xiong Q, Shao G (2015) Vet Microbiol 175:58–67
Li GY, Du KJ, Wang JQ, Liang JW, Kou JF, Hou XJ, Ji LN, Chao H (2013) J Inorg Biochem 119:43–53
Alapetite C, Wachter T, Sage E, Moustacchi E (1996) Int J Radiat Biol 69:359–369
Bartels RH, Van der Linden YM, Van der Graaf WT (2008) CA Cancer J Clin 58:245–259
Shapiro GI, Harper JW (1999) J Clin Invest 104:1645–1653
Wang D, Lippard SJ (2005) Nat Rev Drug Discov 4:307–320
Singh T, Sharma SD, Katiyar SK (2011) PLoS ONE 6:e27444
Crompton M (1999) Biochem J 341:233–249
Halestrap AP, Clarke SJ, Javadov SA (2004) Cardiovasc Res 61:372–385
Laver DR (2007) Biophys J 92:3541–3555
Deretic V (2011) Immunol Rev 240:92–104
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Nature 451:1069–1075
Shintani T, Klionsky DJ (2004) Science 306:990–995
Kondo Y, Kanzawa T, Sawaya R, Kondo S (2005) Nat Rev Cancer 5:726–734
Harikumar KB, Kunnumakkara AB, Sethi G, Diagaradjane P, Anand P, Pandey MK, Gelovani J, Krishnan S, Guha S, Aggarwal BB (2010) Int J Cancer 127:257–268
Schmitz KJ, Ademi C, Bertram S, Schmid KW, Baba HA (2016) World J Surg Oncol 14:189
Maejima Y, Isobe M, Sadoshima J (2016) J Mol Cell Cardiol 95:19–25
Huang X, Bai HM, Chen L, Li B, Lu YC (2010) J Clin Neurosci 17:1515–1519
Apel K, Hirt H (2004) Annu Rev Plant Biol 55:373–399
Khan AU, Wilson T (1995) Chem Biol 2:437–445
Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ (2008) Proc Natl Acad Sci USA 105:11628–11633
Fu Y, Romero MJ, Habtemariam A, Snowden ME, Song LJ, Clarkson GJ, Qamar B, Pizarro AM, Unwin PR, Sadler PJ (2012) Chem Sci 3:2485–2494
Fu Y, Habtemariam A, Pizarro AM, Van Rijt SH, Healey DJ, Cooper PA, Shnyder SD, Clarkson GY, Sadler PJ (2010) J Med Chem 53:8192–8196
Tang B, Wan D, Lai SH, Yang HH, Zhang C, Wang XZ, Zeng CC, Liu YJ (2017) J Inorg Biochem 173:93–104
Wan D, Lai SH, Zeng CC, Zhang C, Tang B, Liu YJ (2017) J Inorg Biochem 173:1–11
Tang B, Han BJ, Wan D, Lai SH, Wang XZ, Zhang C, Zeng CC, Liu YJ (2017) Transit Met Chem 42:373–386
Duriez PJ, Shah GM (1997) Biochem Cell Biol 75:337–349
Taylor RC, Cullen SP, Martin SJ (2008) Nat Rev Mol Cell Biol 9:231–241
Estaquier J, Vallette F, Vayssiere JL, Mignotte B (2012) Adv Exp Med Biol 942:157–183
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Nat Rev Clin Oncol 10:143–153
Acknowledgements
This work was supported by the Natural Science Foundation of Guangdong Province (No. 2016A030313728) and Jiangxi University of Technology for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liang, ZH., Wan, D., Yi, QY. et al. A cyclometalated iridium(III) complex induces apoptosis and autophagy through inhibition of the PI3K/AKT/mTOR pathway. Transit Met Chem 43, 243–257 (2018). https://doi.org/10.1007/s11243-018-0210-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0210-z