
Abstract
WRKY transcription factors play essential roles in mediating various stress responses in plants. Although numbers of researches have investigated the functional mechanisms of WRKY TFs, relative to the research progress in model plants, rather limited numbers of WRKY TFs have been functionally characterized in cotton. In this study, we isolated and characterized GhWRKY44, a group I WRKY gene from cotton (Gossypium hirsutum). Subcellular localization indicated that GhWRKY44 was localized to the nucleus. Additionally, a group of cis-acting elements associated with the response to environmental stresses were predicted in the promoter. The expression of GhWRKY44 can be induced by pathogen injection, abiotic stresses and diverse signaling molecules. Furthermore, the overexpression of GhWRKY44 in N. benthamiana exhibited enhanced resistance to bacterial pathogen R. solanacearum and fungal pathogen R. solani compared with wild-type plants. Importantly, several defense-related genes were induced in the transgenic plants, including PR-1, PR-2, PR-5 and NPR1 for SA signaling, and PR-4 for JA signaling. The transgenic plants also exhibited lower levels of ROS (H2O2 and O2 −) accumulation than wild-type plants following pathogen infection. Taken together, these results suggest that GhWRKY44 positively regulates pathogen induced plant disease resistance, and these findings will expand our knowledge on the functions of WRKY TFs in multiple signal transduction pathways of disease resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pathogen infection is one of the most severe biotic stresses that restrict plant growth and crop production. To reduce the adverse effects of pathogen attack, plants have evolved a variety of efficient defense mechanisms, such as physiological, morphological and biochemical adaptations (Qiu et al. 2007; Zheng et al. 2007; Zhang et al. 2012). Pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) and effector-triggered immunity (ETI) are two interconnected branches innate immune in plants for defense against pathogens (Meng and Zhang 2013). Extensive defense-related genes expression has been associated with PTI and ETI, and appears to be controlled by complex signaling networks (Pieterse et al. 2009; Tsuda and Katagiri 2010). Transcription factor-mediated transcriptional regulation of defense-related genes is a key step in the activation of various plant defense responses (Yu et al. 2001; Dang et al. 2013). Previous studies have shown that transcription factors (TFs), such as WRKY TFs, play crucial roles in both PTI and ETI (Pandey and Somssich 2009).
The WRKY transcription factor can interact with W-box, a common cis-element that is present in the promoter regions of various biotic stress-related genes (Rushton et al. 2010). In plants, WRKY proteins belong to a large family with more than 70 members in Arabidopsis thaliana and more than 100 members in rice (Wu et al. 2005; Zhang and Wang 2005). Recently, a genome-wide analysis of the WRKY gene family in cotton (Gossypium raimondii and Gossypium hirsutum) has been taken, 116 WRKY genes have been identified in G. raimondii and 102 WRKY genes have been cloned in G. hirsutum (Dou et al. 2014). A common property of the WRKY family is their DNA-binding domain, which is composed of a highly conserved peptide sequence (WRKYGQK) at its N-terminal end together with a zinc-finger-like motif (Eulgem et al. 2000). Based on the domain structures, WRKY families are divided into three major groups (I, II, and III) and various subgroups (e.g. IIa, IIb, etc.) (Eulgem et al. 2000).
To date, WRKY TFs appear to function in the regulation of defense responses to biotic and abiotic stresses (Jiang and Deyholos 2009; Rushton et al. 2010; Li et al. 2011; Guo et al. 2011; Shi et al. 2014). Increasing evidences suggest that many WRKY genes can positively or negatively regulate the response to pathogens (Kim et al. 2008; Pandey and Somssich 2009; Lai et al. 2011; Mao et al. 2011; Dang et al. 2013). In Arabidopsis, 49 of 72 AtWRKY genes are differentially regulated by pathogen infection or SA treatment (Dong et al. 2003). AtWRKY70 was a positive regulator of disease resistance to pathogens. Meanwhile, AtWRKY70 has been shown to regulate the cross-talk between SA- and JA-mediated signaling by promoting SA-dependent and suppressing JA-dependent defense responses (Li et al. 2004; Knoth et al. 2007). Overexpression of AtWRKY41 in Arabidopsis exhibited enhanced resistance to Pseudomonas syringae, but increased susceptibility to Erwinia carotovora (Higashi et al. 2008). In pepper, overexpression of CaWRKY40 enhanced resistance to R. solanacearum. In contrast, silencing of CaWRKY40 enhanced susceptibility to R. solanacearum. These results suggest that CaWRKY40 played a positive role in the pathogen infection (Dang et al. 2013). However, CaWRKY58 acts as a transcriptional activator of negative regulators in the resistance to R. solanacearum infection (Wang et al. 2013). Furthermore, WRKY TFs frequently regulate the expression of several defense-related genes, via specific binding to the W-box sequences in the promoter regions, to increase the defense response (Yu et al. 2001). AtWRKY18 increased resistance to defense responses in Arabidopsis and enhanced the expression of defense-related genes, such as PR-1, PR-2 and PR-5 (Chen and Chen 2002). Ectopic overexpression of GhWRKY15 in tobacco plants enhanced the resistance to viral and fungal infections and up regulated the expression of NPR1 (NON-EXPRESSOR OF PR1) and several PR (pathogen-related) genes (Yu et al. 2012). Overall, the above findings indicated that the WRKY TFs play an important role in plant disease resistance.
Cotton (G. hirsutum) is one of the most important fibre and oil crops around the world. Various environmental factors have been shown to inhibit its growth and yield (Sunilkumar et al. 2006). Previous researches have suggested that WRKY TFs were involved in defense-related response (Dong et al. 2003; Eulgem and Somssich 2007; Lai et al. 2011). The study of WRKY TFs in cotton will have great potential to help cotton improving the growth and yield. However, only a small number of WRKY proteins have been characterized in cotton (Yu et al. 2012; Shi et al. 2014; Wang et al. 2014; Yan et al. 2014). In this study, GhWKRY44, a group I WRKY gene from cotton, was isolated and characterized. The GhWKRY44 expression patterns under various biotic and abiotic stresses were investigated. Ectopic expression of GhWKRY44 in N. benthamiana enhanced the plant resistance to bacterial and fungal infections. In addition, GhWKRY44-overexpressing plants increased ability to scavenge reactive oxygen species (ROS). These results suggest that GhWRKY44 may participate in multiple mechanisms by which plants respond to pathogen defense.
Materials and methods
Plant materials, growth conditions and stress treatments
The cotton (G. hirsutum L. cv. Lumian 22) seedlings were grown in a growth chamber at 25 °C with 16 h light/8 h dark cycle (relative humidity of 60–75 %). Seven-day-old cotton seedlings were used for various treatments. Seedlings treated with sterile water were chosen as the control. For the tissue-specific expression analysis, the roots, stems and leaves were harvested from the same plant. For the hormone treatments, the seedling leaves were sprayed with SA (2 mM), MeJA (100 μM), ET (5 mM), 6-BA (20 μM), respectively. For the salt and drought treatments, the seedlings were performed by irrigating the seedlings with NaCl (200 mM) and PEG6000 (15 %). For wounding treatment, the seedling leaves were cut with scissors. For the pathogen treatments, the roots of seedlings were dipped into the bacterial pathogen Ralstonia solanacearum (R. solanacearum) suspensions (OD600 = 0.5–0.8) and the fungal pathogens Rhizoctonia solani (R. solani) conidial suspensions (106 spores/ml), respectively. Each treatment was repeated at three times. All the samples were harvested for RNA extraction.
Nicotiana benthamiana (N. benthamiana) seeds were surface-sterilized and planted on Murashige–Skoog (MS) medium for germination, then transplanted in soil and maintained at 25 °C with a 16 h light/8 h dark cycle. Eight-week-old N. benthamiana seedlings were used for the disease resistance assays.
Cloning and sequence analysis of GhWRKY44
The full-length GhWRKY44 cDNA was amplified by reverse transcription-PCR (RT-PCR) and RACE-PCR (Shi et al. 2011). HiTAIL-PCR was performed to obtain the promoter sequence (Liu and Chen 2007). The primers are shown in Table S1. The PCR products were cloned into the pMD18-T vector (TaKaRa, China) after being purified and transformed into Escherichia coli competent cells (E. coli DH5α) for sequencing.
The GhWRKY44 nucleic acid sequences were translated into amino acids using the DNAman 6.0 program. The amino acids sequences were compared with those in the GenBank using BLAST on the NCBI’s (http://www.ncbi.nlm.nih.gov/) homepage. The phylogenetic tree was constructed in MEGA 5.1 program (Tamura et al. 2011) using the neighbor-joining method.
Vector construction and genetic transformation
The entire GhWRKY44 cDNA was inserted downstream of the Cauliflower Mosaic Virus (CaMV) 35S promoter in the binary expression vector pBI121 via SalI and SacI sites. The recombinant plasmid was then electroporated into Agrobacterium tumefaciens (strain LBA4404), and transformated into N. benthamiana by the leaf disc method (Horsch et al. 1985). The transgenic T3 lines were selected for further studies.
Subcellular localization of the 35S-GhWRKY44::GFP fusion protein
A full-length GhWRKY44 ORF without the termination codon was amplified using the specific primers WG1/WG2 (Table S1). The PCR amplification products and the pBI121-GFP binary vector (driven by the constitutive CaMV35S promoter) were both digested with XholI and XbaI, and the fragment was inserted into the pBI121-GFP to yield the expression vector 35S-GhWRKY44::GFP. The 35S-GFP was used as a control. A. tumefaciens strain GV3101 transformed with 35S-GhWRKY44::GFP or 35S-GFP confusion constructs were grown overnight, pelleted, re-suspended in infiltration buffer (10 mM MES pH 5.7, 10 mM MgCl2, and 200 mM Acetosyringone) and adjusted to the final OD600 of 0.8. Then the mixtures were incubated for 3 h at room temperature. Fully expanded leaves of 5-week-old N. benthamiana were infiltrated with the Agrobacterium suspension using a 1 ml syringe. Leaf samples were collected after 2 days infiltration. Images were performed using a confocal laser scanning microscope (LSM 510 META, ZEISS, Germany).
RNA isolation and quantitative real-time PCR analyses
Total RNA was extracted from cotton seedling leaves by modified CTAB method (Lu et al. 2013). While TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) was used to isolate Total RNA from N. benthamiana leaves according to the manufacturer’s instructions. The first-strand cDNA was synthesized by a reverse transcriptase system (Transgen, Beijing, China). Quantitative RT-PCR was used to analyze the expression levels of GhWRKY44 under different treatments. Fragment of GhWRKY44 gene was amplified using the primer pairs QRT1/QRT2 (Table S1). Quantitative real-time PCR (qRT-PCR) was performed using the SYBR PrimeScript™ RT-PCR Kit (TaKaRa, Dalian, China) on a CFX96TM Real-time System (Bio-Rad, Hercules, CA, USA). The PCR program was as follows: pre-denaturation at 95 °C for 30 s; 40 cycles of 95 °C for 30 s, 55 °C for 15 s and 72 °C for 15 s; and a melt cycle from 65 °C to 95 °C. The polyubiquitin gene (UBI) gene and N. benthamiana β-actin gene was used as the standard control. The results were analysed with the CFX Manager software program (version 1.1) using the 2−ΔΔCt comparative CT method (Livak and Schmittgen 2001). The data were examined by multiple comparisons using one-factor analysis of variance (ANOVA). The significant differences were determined according to Duncan’s multiple range test using Statistical Analysis System (SAS) version 9.1 software.
Pathogen infection
Bacterial and fungal cultures were performed as described previously (Lu et al. 2013). Detached uniform leaves from 8-week-old T3 transgenic and wild-type plants were inoculated with suspensions of R. solanacearum bacteria (OD600 = 0.5–0.8) or R. solani spores (106 spores/ml) using a syringe with a needle. The respective fourth leaves were harvested a transparent culture dish under glasshouse condition for the preparation of RNA or histochemical staining. In addition, the infection was confirmed using the trickle irrigation method to observe disease symptoms (Li et al. 2014). The inoculated plants with roots in soil were kept in a moist chamber at 25 °C for 24 h in the dark and transferred glasshouse condition at 25 °C. Bacterial growth was monitored by performing serial dilutions onto King’s B agar medium. The disease resistance analysis was repeated three times.
3,3′-Diaminobenzidine (DAB), nitro blue tetrazolium (NBT), and trypan blue staining assays
Uniform leaves form 8-week-old T3 transgenic and wild-type plants were inoculated by R. solanacearum bacteria (OD600 = 0.5–0.8) and R. solani spores (106 spores/ml) for staining assays. Hydrogen peroxide (H2O2) was detected in the leaves of the plants using DAB staining. The infection leaves were incubated in a DAB solution (1 mg/ml, pH 3.8) for 12 h at 25 °C in the dark. After staining, the leaves were soaked overnight in 95 % ethanol to remove chlorophyll (Thordal-Christensen et al. 1997). Superoxide anion (O2 −) was measured in the leaves of the plants via NBT staining. The infection leaves were incubated in a NBT solution (0.1 mg/ml) for 24 h at 25 °C in the dark. After staining, the leaves were also destained overnight in 95 % ethanol (Jabs et al. 1996). The cell death was detected in the leaves using trypan blue staining. After treatment, leaves were stained with lactophenol-trypan blue solution for 5 min, soaked in chloral hydrate (2.5 g/ml) for 12 h to remove chlorophyll (Zhang et al. 2012).
Results
Sequence analysis of GhWRKY44
Based on the vital function of the WRKY motif in the activity of the WRKY family members, we designed pair primers to isolate a cDNA fragment from cotton cotyledons. A predicted full-length cDNA sequence of WRKY gene was successfully obtained by the homology cloning strategy. The deduced full-length sequence of cDNA fragment consisted of 1,716 nucleotides, including a 188 bp 5′-untranslated region (UTR), a 127 bp 3′-UTR, and a 1,401 bp open reading frame (ORF). The cDNA encodes a polypeptide of 467 amino acids with a putative molecular weight of 51.39 kDa and an isoelectric point of 8.76. The putative WRKY clone from cotton exhibits high sequence similarity to Arabidopsis thaliana AtWRKY44 (NM_129282), so the cotton WRKY gene was designated as GhWRKY44 (GenBank accession no. KJ871340).
Alignment analysis of GhWRKY44 protein sequence with related sequences revealed that it shared 45.1, 97.4, 60.54, 42.3, 48.5 and 58.6 % identity with AtWRKY44 (NP_001078015), GhWRKY97(AIE43909), GmWRKY44(XP_003520637), BnWRKY44 (ACH99805), CsWRKY44 (NP_001267637) and MtWRKY44 (XP_003625561), respectively (Fig. 1a). Similar to the other members of WRKY group I, the putative GhWRKY44 protein contains two typical DNA binding domain that possess the highly conserved amino acid sequence WRKYGQK and two putative zinc finger motifs (C–X4–C–X22–23–H–X1–H) (Fig. 1a). It suggests that the GhWRKY44 represent a member of WRKY group I. A phylogenetic tree was also constructed to determine the evolutionary relationship between GhWRKY44 and the other WRKY proteins (Fig. 1b). Following the standard of previous classification, GhWRKY44 protein was closely related to group I WRKY family members, including AtWRKY44, GhWRKY44-like, GhWRKY97, GrWRKY98, BnWRKY44 and GrWRKY97. These results strongly indicate that GhWRKY44 belongs to group I WRKY family.

Characterization and sequence analysis of GhWRKY44. a Sequence alignment of the deduced GhWRKY44 protein with AtWRKY44 (NP_001078015), GhWRKY97 (AIE43909), GmWRKY44 (XP_003520637), BnWRKY44 (ACH99805), CsWRKY44 (NP_001267637) and MtWRKY44 (XP_003625561). Identical and similar amino acids were shaded in dark blue and pink, respectively. The WRKY domains are shown in frames. The cysteine and histidine residues of the putative zinc finger motif were indicated by arrowheads (▼). The putative NLS was indicated by straight line. b Phylogenetic analysis of GhWRKY44. The phylogenetic tree was constructed using the software MEGA 5.1. Neighbor-joining method was used and bootstrap analysis was performed with 1,000 replications. Bootstrap values above 50 were indicated at branch points. The species of origin is indicated by the abbreviation before the gene names: At, Arabidopsis thaliana; Bn, Brassica napus; Gh, Gossypium hirsutum; Gr, Gossypium raimondii. (Color figure online)
Subcellular localization of GhWRKY44
Using a prediction program for protein subcellular localization, PSORT (http://psort.ims.u-tokyo.ac.jp), it was predicted that GhWRKY44 might be localized in the nucleus due to the existence of the putative nuclear localization signal (KRRK). To further confirm this prediction, we constructed the 35S-GhWRKY44::GFP overexpression vector driven by the constitutive CaMV35S promoter. As shown in Fig. 2b, the tobacco leaf cell carrying the 35S-GhWRKY44::GFP emitted fluorescence only in nucleus, whereas 35S::GFP control were present in multiple subcellular compartments, including the cytoplasm and nuclei. These results indicate that the GhWRKY44 is a nuclear protein, which might function in the nucleus.

Subcellular localization of GhWRKY44 fusion protein in tobacco leaf epidermal cells. a Schematic diagram of the 35S-GhWRKY44::GFP fusion construct and the 35S-GFP construct. b Transient expression of the 35S-GhWRKY44::GFP and 35S-GFP constructs in tobacco leaf epidermal cells
Analysis of partial putative cis-acting elements in the 5′-flanking region of GhWRKY44
To elucidate the mechanism underlying the expression patterns of GhWRKY44, a 1,418 bp fragment of 5′-flanking region of GhWRKY44 was obtained by hiTAIL-PCR method. The Plant CARE database (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) was used to analyze the putative cis-acting elements of this region. All of the identified cis-elements are shown in Fig. 3. Several putative cis-acting elements may be involved in defense responses, including CGTCA-motif involved in the MeJA-responsiveness, TCA-element involved in salicylic acid responsiveness, TC-rich repeats involved in defense and stress responsiveness and ethylene-responsive element ERE. In addition, abiotic stress responsive cis-acting elements were also identified, such as the HSE element which involved in heat stress responsive, MBS representing the MYB binding site involved in drought-inducibility, gibberellin-responsive element GARE-motif and so on. These data suggest that GhWRKY44 may play a role in the response to various environmental stresses.

Nucleotide sequence of the promoter of GhWRKY44. The predicted transcription initiation site is highlighted in grey (C). The start codon is marked with an asterisk, and the putative core promoter consensus sequences (TATA-box and CAAT-box) are highlighted in grey. The W-box and putative cis-acting elements are indicated by boxes and their corresponding names are given above each element. Arrows indicate the direction of the cis-acting elements
Expression patterns of GhWRKY44 mRNA
To further identify the expression patterns of GhWRKY44 gene, quantitative RT-PCR was performed using 7-day-old cotton seedlings. As shown in Fig. 4a, the expression of GhWRKY44 was higher in leaves than that in roots and stems.

Expression patterns of GhWRKY44 in different tissues and under different stress conditions. a Tissue-specific expression of GhWRKY44 was detected with quantitative RT-PCR. Total RNA were extracted from roots, stems and cotyledon leaves of 7-day-old cotton seedlings. b–j The expression patterns of GhWRKY44 in cotton seedlings were analysed with quantitative RT-PCR following treatments with R. solanacearum (b), R. solani (c), wounding (d),15 % PEG6000 (e), 200 mM NaCl (f), 2 mM SA (g), 5 mM ET (h), 100 μM MeJA (i), 20 μM 6-BA (j). The lanes represent various time points during the treatments. The ubiquitin gene was used as an internal control. Different letters above the columns indicate significant differences (P < 0.01) as determined by Duncan’s multiple range test. Each experiment was repeated at least three times
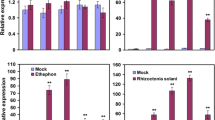
To explore whether GhWRKY44 expression is triggered by biotic stresses, the cotton seedlings were infected with R. solanacearum and R. solani. As shown in Fig. 4b, GhWRKY44 transcripts was induced in 1d and noticeably elevated on 2d after R. solanacearum treatment. Under R. solani treatment, the expression of GhWRKY44 was rapidly enhanced in 1d, then decreasing gradually to the level of the control (Fig. 4c). These results indicated that GhWRKY44 might play crucial roles in the plant pathogen defense response.
In addition, we also examined the expression of GhWRKY44 under abiotic stresses. For wounding treatment, the expression of GhWRKY44 was induced and reached maximal level at 2 h (Fig. 4d). As shown in Fig. 4e, the transcript level of GhWRKY44 was increased at 2–12 h after PEG6000 treatment, with a highest level at 2 h. After NaCl treatment, a slight increase of GhWRKY44 transcripts appeared at 4 h, while the induction phenomenon is not obvious (Fig. 4f). The drought and salinity results fit well with earlier research results of GhWRKY97 (Dou et al. 2014). These results showed that the expression levels of GhWRKY44 can be regulated by abiotic stresses.
To explore the molecular mechanism underlying the responsiveness to various stresses, cotton seedlings were inoculated with signaling molecules, including SA, ET, MeJA and 6-BA. As shown in Fig. 4g, h, the expression levels of GhWRKY44 were drastically elevated after SA and ET treatments at the same time point. However, the transcript level of GhWRKY44 was slightly induced through MeJA treatment (Fig. 4i). For 6-BA treatment, the GhWRKY44 expression was also increased (Fig. 4g). Based on these results, we deduced that GhWRKY44 might be involved in complex signaling pathways and played crucial roles in controlling the expression of downstream defense genes.
Overexpression of GhWRKY44 exhibit enhanced pathogen resistance
The characterization of promoter and endogenous expression implied that the GhWRKY44 might be involved in plant defense responses. In order to further analyze the putative function of GhWRKY44 in plant, full-length GhWRKY44 was cloned into the binary vector pBI121 under control of the CaMV35S promoter and transformed into N. benthamiana. In all, 14 independent transgenic lines were obtained by kanamycin resistance selection (data not shown). The expression levels in different lines (10 randomly selected lines) were detected using semi-quantitative RT-PCR (Fig. 5a). Compared to the control, no obvious morphological changes were observed in the transgenic lines (Fig. 5b). However, transgenic lines accumulated different levels of GhWRKY44 transcripts compared with the wild-type plants. Three representative lines, OE1 (5#), OE2 (8#), and OE3 (9#), exhibiting different expression levels of T3 progeny were selected for further investigation.

Identification of transgenic plants and bacterial pathogen resistance analysis. a The expression levels of GhWRKY44 in the T1 progeny of transgenic plants. b The developmental phenotype from 8-week-old T3 generation transgenic plants. c Disease signs on the detached leaves of wild-type and transgenic plants 6 days after R. solanacearum infection. The cell death was visualized by trypan blue staining. d The lesion diameters on the leaves after inoculation with R. solanacearum. e The phenotypes of the wild-type and transgenic plants after inoculation with R. solanacearum using the trickle irrigation method. f The survival rates of wild-type and transgenic plants after inoculation with R. solanacearum. Different letters above the columns indicate significant differences (P < 0.05) as determined by Duncan’s multiple range test. Each experiment was repeated at least three times
Eight-week-old transgenic plants were used for the resistance tests. When the detached leaves were inoculated with R. solanacearum over 6 days, the leaves of the transgenic lines exhibited more resistance than the wild-type plants. It was found that the transgenic lines showed minor signs of chlorosis and relatively smaller lesion diameters than those of the wild-type plants (Fig. 5c, d). Trypan blue staining showed relatively less staining in the leaves of the transgenic plants than those of the wild-type plants, which indicated that overexpression of GhWRKY44 decreased the levels of cell death in the transgenic plants (Fig. 5c). The trickle irrigation method was also used to examine the response to the bacterial pathogen. Marked wilting symptoms were observed on the wild-type plants, whereas the transgenic lines exhibited only slight wilting symptoms after 10 days inoculation. In addition, the transgenic lines showed a relatively higher survival rate than the wild-type plants (Fig. 5e, f). These results indicated that GhWRKY44 could enhance resistance to the bacterial pathogen in transgenic plants.
To investigate whether transgenic plants possess enhanced fungal resistance, 8-week-old plants were inoculated with R. solani. Conidial suspensions spores (106 spores/ml) were sprayed on detached leaves. After 7 days inoculation, the increased disease symptoms characterized by extensive chlorosis and many more spores were present in the wild-type leaves than the transgenic leaves (Fig. 6a, b). In addition, Trypan blue staining revealed that the wild-type leaves showed a more serious cell death phenotype than the transgenic lines leaves. Similarly, the trickle irrigation method was also used to examine the response to the fungal pathogen. After 2-week inoculation, the transgenic lines displayed less severe disease symptoms. The wilting symptoms on the transgenic plants were slighter than those on the wild-type leaves, with a higher survival rate than the wild-type plants (Fig. 6c, d). These results demonstrated that GhWRKY44 may be a positive regulator of plant defense against the fungal pathogens.

Enhanced resistance of GhWRKY44-overexpressing plants to fungi. a Disease signs on the detached leaves of wild-type and transgenic plants 7 days after inoculation with R. solani. The cell death was visualized by trypan blue staining. b The number of spores per cotyledon at 7 days after R. solani infection. c The symptoms of wild-type and transgenic plants inoculated with R. solani using the trickle irrigation method. d The survival rates of wild-type and transgenic plants after inoculation with R. solani. Different letters above the columns indicate significant differences (P < 0.05) as determined by Duncan’s multiple range test. Each experiment was repeated at least three times
GhWRKY44 regulates the accumulation of ROS
Generally, pathogens invasion in plant often result in the generation of ROS, including hydrogen peroxide (H2O2), singlet oxygen, superoxide anion (O2 −), and hydroxyl radicals (Hernández et al. 2004; Yoshioka et al. 2003). To test whether ROS accumulation was related to the pathogens invasion of the transgenic plants, the accumulation of H2O2 and O2 − were monitored in transgenic and wild-type plants following exposure to R. solanacearum or R. solani. The accumulation of H2O2 and O2 − were detected by 3,3′-diaminobenzidine (DAB) and nitro blue tetrazolium (NBT) staining methods, respectively. As shown in Fig. 7a, following exposure to R. solanacearum or R. solani, the DAB staining was found to be much lighter in the leaves of the transgenic plants, indicating that lower levels accumulation of H2O2 compared with wild-type plants. Similar to that observed in DAB staining, NBT staining revealed the leaves of the transgenic plants also accumulated less O2 − compared with wild-type plants (Fig. 7b). These results indicated that overexpression of GhWRKY44 could enhance defense resistance by inhibiting the accumulation of pathogen-induced ROS.

Analysis of ROS accumulation in wild type and transgenic plants in response to pathogens infection. a The accumulation of H2O2 using DAB staining after inoculation with R. solanacearum and R. solani. b The accumulation of O2 − using NBT staining after inoculation with R. solanacearum and R. solani
Overexpression of GhWRKY44 activates the expression of defense-related genes
Generally, the expression levels of PR genes and NPR1 gene, are involved in the defense pathway, such as salicylic acid (SA) and jasmonic acid (JA) signaling pathways (Agrawal et al. 2000; Yu et al. 2001; Pieterse and Van Loon 2004). To explore the possible mechanisms underlying the enhanced pathogen resistance in the transgenic plants, we characterized the expression levels of several PR genes and NPR1 gene before and after infection with R. solanacearum using qRT-PCR. As shown in Fig. 8a, the expression of PR2 (β-1,3-glucanase) gene was greater in the three transgenic lines than the wild-type plants before infection with R. solanacearum. To our surprise, other defense-related genes, including PR1a, PR4, PR5 and the NPR1 showed lower expression levels in the transgenic plants compared with the wild-type plants (Fig. 8a). After infection with R. solanacearum, the transcriptional levels of these defense-related genes were all up regulated in transgenic plants (Fig. 8b). Furthermore, the expression of PR2, PR4 and NPR1 genes was much higher than the wild-type plants after infection with R. solanacearum. Notably, PR1a, PR2, PR5 and NPR1 are SA signaling-related genes, and PR4 is JA signaling-related gene. Thus, it was speculated that the enhanced defense resistance conferred by GhWRKY44 in transgenic plants might be related to SA- and JA dependent signaling pathway.

Expression of defense-related genes in wild-type and transgenic plants by qRT-PCR. a The expression levels of defense related genes under normal conditions. b The expression levels of defense related genes after inoculation with R. solanacearum. Different letters for each defense related gene indicate significant differences (P < 0.05) as determined by Duncan’s multiple range test
Discussion
The transcriptional regulation of plant defensive genes plays a pivotal role in the activation of plant defense responses (Jing et al. 2009). The significance of WRKY TFs in response to various biotic and abiotic stresses had been described (Eulgem and Somssich 2007; Rushton et al. 2010; Chen et al. 2012). Recently, a large body of evidences suggested that many WRKY genes have been implicated in the regulation of pathogens infection (Oh et al. 2008; Cai et al. 2008; Yu et al. 2012; Shimono et al. 2012). To explore the function of the WRKY transcription factors in cotton, a group I WRKY gene named GhWRKY44 was isolated from G. hirsutum and the 5′-flanking region containing two W-box were identified. W-box is the minimal core element necessary for binding of a WRKY protein to DNA (Rushton et al. 1996, Ciolkowski et al. 2008). And W-boxes can be found in the promoters systemic acquired resistance related (SAR) genes, including non-expresser of PR genes 1, and pathogenesis related 1 (Fu and Dong. 2013). Also, several other putative cis-elements involved in defense response including MeJA, SA, ET were found. These results indicated that GhWRKY44 may play positive roles in disease defense.
In this study, the GhWRKY44 transcriptions were induced by diverse signaling molecules and pathogens (Fig. 4). So we speculated that GhWRKY44 might be involved in defense responses. To test the role of GhWRKY44 in pathogen infection, transgenic plants were inoculated with bacterial and fungal pathogens, respectively. The overexpression of GhWRKY44 enhanced the resistance to bacterial pathogen R. solanacearum and fungal pathogen R. solani in transgenic plants (Fig. 5, 6). Moreover, the expression of defense related genes showed higher level in transgenic plants than wild-type plants (Fig. 8). These results strongly suggest that GhWRKY44 functions as a positive regulator to pathogen resistance in transgenic plants. Most of the WRKY TFs in plants reported recently have shown positive roles in pathogen resistance (Zheng et al. 2006; Liu et al. 2007; Peng et al. 2012; Yu et al. 2012; Shi et al. 2014). For example, loss-of-function mutants of AtWRKY33 caused enhanced susceptibility to the necrotrophic fungal pathogens Botrytis cinerea and Alternaria brassicicola, while transgenic overexpression of AtWRKY33 increased resistance to the two neurotrophic fungal pathogens (Zheng et al. 2006). In rice, OsWRKY45 could positive regulate the disease resistance to rice blast fungus Magnaporthe grisea by mediating a signaling pathway downstream of SA (Shimono et al. 2007). These results provide further evidence for WRKY TFs playing positive roles in regulating disease resistance.
Mounting evidences suggest that plants challenged with pathogens are often exposed to the accumulation of ROS (Kotchoni and Gachomo 2006). It is critical to modulate ROS to an appropriate level. Low ROS concentrations can act as signaling molecules to regulate cell growth, while high ROS concentrations induce cellular senescence and lead to oxidative damage (Poli et al. 2004). The overexpression of GhWRKY44 in transgenic plants resulted in the decreased accumulation of H2O2 and O2 − than wild-type plants under pathogen infection (Fig. 7). These results suggest that the enhanced resistance to pathogen infection in GhWRKY44-overexpressing plants may be associated with the removal of ROS. Recently, it was also reported that overexpression of GhWRKY39 in N. benthamiana enhanced tolerance to pathogen infection and decreased H2O2 accumulation than wild-type plants (Shi et al. 2014). Overall, WRKY TFs may play crucial roles in ROS homeostasis, and alleviated the ROS-associated oxidative injury.
SA and JA/ET are two well-studied signaling pathways which play important roles in the regulation of plant defense responses to pathogen infection (Spoel and Dong 2008; Peng et al. 2012). Recent studies have shown that a variety of WRKY genes are involved in different defense signaling pathways (Birkenbihl et al. 2012; Peng et al. 2012; Dang et al. 2013). In Arabidopsis, WRKY25 functions as a negative regulator of SA-mediated defense responses to P. syringae by T-DNA insertion mutants and transgenic overexpression methods (Zheng et al. 2007). In rice, JA plays an important role in WRKY30-mediated defense responses to fungal pathogens (Peng et al. 2012). In this study, GhWRKY44 could be induced by SA, MeJA and ET, which suggested that GhWRKY44 might be involved in SA or JA/ET signaling pathway. In addition, the expression of the marker genes for different signaling pathways (PR1a, PR2, PR5 and NPR1 for SA signaling; PR4 for JA signaling) were significantly increased in GhWRKY44-overexpressing plants under R. solanacearum infection (Fig. 8). Similarly, overexpression of OsWRKY03 enhanced the resistance to bacterial blight pathogen, and induced the expression of several pathogenesis-related genes in transgenic plants. Overall, OsWRKY03 is located upstream of OsNPR1 as a transcriptional activator in SA-dependent or JA-dependent defense signaling pathways (Liu et al. 2005). Thus, it is reasonable to speculate that GhWRKY44 may be involved in the crosstalk between the SA- and JA-mediated pathogen defense pathways.
In conclusion, we isolated and characterized a group I WRKY member, GhWRKY44, in cotton. The results of the present study strongly suggest that overexpression of GhWRKY44 in N. benthamiana enhanced their resistance to bacterial and fungal pathogens. The enhanced resistance may be associated with the SA-, JA- and ROS-mediated defense mechanism. The study provides evidence for the positive regulatory functions of GhWRKY44 resistance against pathogenic infection. Further studies on the function of GhWRKY44 in transgenic cotton should be elucidated and loss-of function data in cotton are particularly needed.
Abbreviations
- CTAB:
-
Cetyltrimethyl ammonium bromide
- DAB:
-
3,3′-Diaminobenzidine
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- ET:
-
Ethylene
- GFP:
-
Green fluorescent protein
- Gh:
-
Gossypium hirsutum
- hiTAIL-PCR:
-
High-efficiency TAIL-PCR
- MeJA:
-
Methyl jasmonate
- MES:
-
2-(N-Morpholino)ethanesulfonic acid
- MS medium:
-
Murashige and Skoog medium
- Nb:
-
Nicotiana benthamiana
- NBT:
-
Nitro blue tetrazolium
- NPR1:
-
Non-expression of PR1
- OE:
-
Overexpression
- ORF:
-
Open reading frame
- PR:
-
Pathogenesis-related
- RACE:
-
Rapid amplification of cDNA ends
- ROS:
-
Reactive oxygen species
- RT-PCR:
-
Reverse transcription-PCR
- R. solanacearum :
-
Ralstonia solanacearum
- R. solani :
-
Rhizoctonia solani
- qRT-PCR:
-
Quantitative real-time PCR PCR
- SA:
-
Salicylic acid
References
Agrawal GK, Jwa NS, Rakwal R (2000) A novel rice (Oryza sativa L.) acidic PR1 gene highly responsive to cut, phytohormones, and protein phosphatase inhibitors. Biochem Biophys Res Commun 274:157–165
Birkenbihl RP, Diezel C, Somssich IE (2012) Arabidopsis WRKY33 is a key transcriptional regulator of hormonal and metabolic responses toward Botrytis cinerea infection. Plant Physiol 159:266–285
Cai M, Qiu D, Yuan T, Ding X, Li H, Duan L, Xu C, Li X, Wang S (2008) Identification of novel pathogen-responsive cis-elements and their binding proteins in the promoter of OsWRKY13, a gene regulating rice disease resistance. Plant, Cell Environ 31:86–96
Chen C, Chen Z (2002) Potentiation of developmentally regulated defense response by AtWRKY18, a pathogen-induced Arabidopsis transcription factor. Plant Physiol 129:706–716
Chen L, Song Y, Li S, Zhang L, Zou C, Yu D (2012) The role of WRKY TFs in plant abiotic stresses. Biochim Biophys Acta 1819:120–128
Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE (2008) Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol Biol 68(1–2):81–92
Dang FF, Wang YN, Yu L, Eulgem T, Lai Y, Liu ZQ, Wang X, Qiu AL, Zhang TX, Lin J, Chen YS, Guan DY, Cai HY, Mou SL, He SL (2013) CaWRKY40, a WRKY protein of pepper, plays an important role in the regulation of tolerance to heat stress and resistance to Ralstonia solanacearum infection. Plant, Cell Environ 36:757–774
Dong J, Chen C, Chen Z (2003) Expression profile of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol 51:21–37
Dou LL, Zhang XH, Pang CY, Song MZ, Wei HL, Fan SL, Yu SX (2014) Genome wide analysis of the WRKY gene family in cotton. Mol Genet Genomics 289(6):1103–1121
Eulgem T, Somssich IE (2007) Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol 10:366–371
Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5:199–206
Fu ZQ, Dong X (2013) Systemic acquired resistance: turning local infection into global defense. Annu Rev Plant Biol 64:839–863
Guo RY, Yu FF, Gao Z, An HL, Cao XC, Guo XQ (2011) GhWRKY3, a novel cotton (Gossypium hirsutum L.) WRKY gene, is involved in diverse stress responses. Mol Biol Rep 38:49–58
Hernández JA, Rubio M, Olmos E, Ros-Barceló A, Martínez-Gómez P (2004) Oxidative stress induced by long-term PlumPox virus infection in peach (Prunus persica). Physiol Plant 122:486–495
Higashi K, Ishiga Y, Inagaki Y, Toyoda K, Shiraishi T, Ichinose Y (2008) Modulation of defense signal transduction by flagellin-induced WRKY41 transcription factor in Arabidopsis thaliana. Mol Genet Genomics 279:303–312
Horsch RB, Fry JE, HoVmann NL, Eichholtz D, Eichholtz D, Rogers SA, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227:1229–1231
Jabs T, Dietrich RA, Dangl JL (1996) Initiation of runaway cell death in an Arabidopsis mutant by extracellular superoxide. Science 273:1853–1856
Jiang Y, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69:91–105
Jing S, Zhou X, Song Y, Yu D (2009) Heterologous expression of OsWRKY23 gene enhances pathogen defense and dark-induced leaf senescence in Arabidopsis. Plant Growth Regul 58:181–190
Kim KC, Lai Z, Fan B, Chen Z (2008) Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone dea-cetylase 19 in basal defense. Plant Cell 20:2357–2371
Knoth C, Ringler J, Dangl JL, Eulgem T (2007) Arabidopsis WRKY70 is required for full RPP4-mediated disease resistance and basal defense against Hyaloperonospora parasitica. Mol Plant Microbe Interact 20:120–128
Kotchoni SO, Gachomo EW (2006) The reactive oxygen species network pathways: an essential prerequisite for perception of pathogen attack and the acquired disease resistance in plants. J Biosci 31:389–404
Lai Z, Li Y, Wang F, Cheng Y, Fan B, Yu JQ, Chen Z (2011) Arabidopsis sigma factor binding proteins are activators of the WRKY33 transcription factor in plant defense. Plant Cell 23:3824–3841
Li J, Brader G, Palva ET (2004) The WRKY70 transcription factor: a node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense. Plant Cell 16:319–331
Li S, Fu Q, Chen L, Huang W, Yu D (2011) Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 233:1237–1252
Li YZ, Zhang L, Lu WJ, Wang XL, Wu CA, Guo XQ (2014) Overexpression of cotton GhMKK4 enhances disease susceptibility and affects abscisic acid, gibberellin and hydrogen peroxide signalling in transgenic Nicotiana benthamiana. Mol Plant Pathol 15(1):94–108
Liu YG, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechnique 43:649–656
Liu XQ, Bai XQ, Qian Q, Wang XJ, Chen MS, Chu CC (2005) OsWRKY03, a rice transcriptional activator that functions in defense signaling pathway upstream of OsNPR1. Cell Res 15:593–603
Liu X, Bai X, Wang X, Chu C (2007) OsWRKY71, a rice transcription factor, is involved in rice defense response. J Plant Physiol 164:969–979
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using realtime quantitative PCR and the 2−ΔΔC(T) Method. Methods 25(4):402–408
Lu WJ, Chu XQ, Li YZ, Wang C, Guo XQ (2013) Cotton GhMKK1 induces the tolerance of salt and drought stress, and mediates defense responses to pathogen infection in transgenic Nicotiana benthamiana. PLoS ONE 8:e68503
Mao G, Meng X, Liu Y, Zheng Z, Chen Z, Zhang S (2011) Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell 23:1639–1653
Meng XZ, Zhang SQ (2013) MAPK Cascades in Plant Disease Resistance Signaling. Annu Rev Phytopathol 51:245–266
Oh SK, Baek KH, Park JM, Yi SY, Yu SH, Kamoun S, Choi D (2008) Capsicum annuum WRKY protein CaWRKY1 is a negative regulator of pathogen defense. New Phytol 177:977–989
Pandey SP, Somssich IE (2009) The role of WRKY transcription factors in plant immunity. Plant Physiol 150:1648–1655
Peng X, Hu Y, Tang X, Zhou P, Deng X, Wang H, Guo Z (2012) Constitutive expression of rice WRKY30 gene increases the endogenous jasmonic acid accumulation, PR gene expression and resistance to fungal pathogens in rice. Planta 236:1485–1498
Pieterse CM, Van Loon LC (2004) NPR1: the spider in the web of induced resistance signaling pathways. Curr Opin Plant Biol 7:456–464
Pieterse CMJ, Leon-Reyes A, Van der Ent S, Van Wees SCM (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5:308–316
Poli G, Leonarduzzi G, Biasi F, Chiarpotto E (2004) Oxidative stress and cell signalling. Curr Med Chem 11:1163–1182
Qiu D, Xiao J, Ding X, Xiong M, Cai M, Cao Y, Li X, Xu C, Wang S (2007) OsWRKY13 mediates rice disease resistance by regulating defense-related genes in salicylate- and jasmonate-dependent signaling. Mol Plant Microbe Interact 20:492–499
Rushton PJ, Torres JT, Parniske M, Wernert P, Hahlbrock K, Somssich IE (1996) Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J 15(20):5690–5700
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258
Shi J, Zhang L, An HL, Wu CA, Guo XQ (2011) GhMPK16, a novel stress-responsive group D MAPK gene from cotton, is involved in disease resistance and drought sensitivity. BMC Mol Biol 12:22
Shi WN, Liu DD, Hao LL, Wu CA, Guo XQ, Li H (2014) GhWRKY39, a member of the WRKY transcription factor family in cotton, has a positive role in disease resistance and salt stress tolerance. Plant Cell, Tissue Organ Cult 118:17–32
Shimono M, Sugano S, Nakayama A, Jiang CJ, Ono K, Toki S, Takatsuji H (2007) Rice WRKY45 plays a crucial role inbenzothiadiazole-inducible blast resistance. Plant Cell 19:2064–2076
Shimono M, Koga H, Akagi A, Hayashi N, Goto S, Sawada M, Kurihara T, Matsushita A, Sugano S, Jiang CJ, Kaku H, Inoue H, Takatsuji H (2012) Rice WRKY45 plays important roles in fungal and bacterial disease resistance. Mol Plant Pathol 13:83–94
Spoel SH, Dong X (2008) Making sense of hormone crosstalk during plant immune responses. Cell Host Microbe 3:348–351
Sunilkumar G, Campbell LM, Puckhaber L, Stipanovic RD, Rathore KS (2006) Engineering cottonseed for use in human nutrition by tissue-specific reduction of toxic gossypol. Proc Natl Acad Sci USA 103:18054–18059
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thordal-Christensen H, Zhang ZG, Wei YD, Collinge DB (1997) Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersen-sitive response during the barley-powdery mildew interaction. Plant J 11:1187–1194
Tsuda K, Katagiri F (2010) Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr Opin Plant Biol 13:459–465
Wang Y, Dang F, Liu Z, Wang X, Eulgem T, Lai Y, Yu L, She J, Shi Y, Lin J, Chen C, Guan D, Qiu A, He S (2013) CaWRKY58, encoding a group I WRKY transcription factor of Capsicum annuum, negatively regulates resistance to Ralstonia solanacearum infection. Mol Plant Pathol 14(2):131–144. doi:10.1111/j.1364-3703.2012.00836.x
Wang XL, Yan Y, Li YZ, Chu XA, Wu CQ, Guo XQ (2014) GhWRKY40, a multiple stress-responsive cotton WRKY gene, plays an important role in the wounding response and enhances susceptibility to Ralstonia solanacearum infection in transgenic Nicotiana benthamiana. PLoS One 18(9):e93577
Wu KL, Guo ZJ, Wang HH, Li J (2005) The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res 12:9–26
Yan HR, Jia HH, Chen XB, Hao LL, An HL, Guo XQ (2014) The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol. doi:10.1093/pcp/pcu133
Yoshioka H, Numata N, Nakajima K, Katou S, Kawakita K, Rowland O, Jones JD, Doke N (2003) Nicotiana benthamiana gp91phox homologs NbrbohA and NbrbohB participate in H2O2 accumulation and resistance to Phytophthora infestans. Plant Cell 15:706–718
Yu D, Chen C, Chen Z (2001) Evidence for an important role of WRKY DNA binding proteins in the regulation of NPR1 gene expression. Plant Cell 13:1527–1540
Yu FF, Huaxia YF, Lu WJ, Wu C, Guo XQ (2012) GhWRKY15, a member of the WRKY transcription factor family identified from cotton (Gossypium hirsutum L.), is involved in disease resistance and plant development. BMC Plant Biol 12:144
Zhang Y, Wang L (2005) The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol 5:1
Zhang L, Li YZ, Lu WJ, Meng F, Wu CA, Guo X (2012) Cotton GhMKK5 affects disease resistance, induces HR-like cell death, and reduces the tolerance to salt and drought stress in transgenic Nicotiana benthamiana. J Exp Bot 63:3935–3951
Zheng Z, Qamar SA, Chen Z, Mengiste T (2006) Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J 48:592–605
Zheng Z, Mosher SL, Fan B, Klessig DF, Chen Z (2007) Functional analysis of Arabidopsis WRKY25 transcription factor in plant defense against Pseudomonas syringae. BMC Plant Biol 10(7):2
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (No. 31171837) and Open Funding Project of State Key Laboratory of Crop Biology, China (No. 2013KF09).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jing Li and Ji Wang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, J., Wang, J., Wang, N. et al. GhWRKY44, a WRKY transcription factor of cotton, mediates defense responses to pathogen infection in transgenic Nicotiana benthamiana . Plant Cell Tiss Organ Cult 121, 127–140 (2015). https://doi.org/10.1007/s11240-014-0688-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0688-9