
Abstract
Cryopreservation of mint shoot tips grown in vitro (Mentha × piperita) was performed after encapsulation in alginate beads. Encapsulated shoot tips were first precultured in sucrose enriched medium (0.75 M) and then dried under a sterile air flow (0–6 h). After cooling in liquid nitrogen and warming in a warm water bath, alginate beads were transferred to solid culture medium for 4 weeks. The effect of dehydration time of the encapsulated shoots was evaluated for water content, cooling and warming rates, ice crystal formation and cellular vitrification, by using low temperature scanning electron microscopy and differential scanning calorimetry. Viability of the recovered material showed a close relation between the dehydration time, cooling and warming rates, ice formation avoidance and tissue vitrification. At short drying periods (up to 3 h), ice crystals were formed and the viability was low or absent. After longer drying periods (5 and 6 h), both beads and specimens became vitrified.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seeds and vegetative material of cultivated mint and its related wild species are susceptible to cryopreservation. Initial studies on mint were mainly focused to its chemical composition, cultivation systems, essential oil extraction and product quality. In the last two decades, the preservation of mint genetic resources, especially long-term, got a lot of attention as a result of growing interest in cultivation of medicinal and aromatic plants. Mint belongs to the genus Mentha L. (Lamiaceae), is native to mild climate areas but has a global range (Towill 2002).
To avoid somaclonal variations that may occur in tissue cultivation and preservation, cryopreservation offers long-term storage possibilities, high stability of phenotypic and genotypic characteristics of stored germplasm, requiring minimal storage space and maintenance; hence has been considered to be an ideal mean for long-term conservation of genetic resources of vegetatively propagated plants (Engelmann 1997; Engelmann and Takagi 2000; Shibli 2000). It is performed in liquid nitrogen (LN) at −196 °C, and it is the only method currently available ensuring the safe, efficient, and cost-effective storage of germplasm of many plant species. Cryopreservation results in arrested metabolic and biochemical processes, such as cell division and growth (Towill 1996; Engelmann and Takagi 2000). Biophysical processes leading to protein and nucleic acid degradation would also be halted. Thus, the plant material can be stored without deterioration or modification for unlimited periods (Ashmore 1997), maintaining its genetic stability and regeneration potential (Rajasekaran 1996; Matsumoto et al. 1998; Towill 2002).
To be successful, cryopreservation must avoid the formation of ice crystals inside cells during immersion in LN. This is achieved by reducing cell water content prior to rapid cooling. Therefore, treatments resulting in low cell water content without causing cell death are key factors for successful cryopreservation (Matsumoto et al. 1998). Several pre-treatments, including cold acclimation, exposure to abscisic acid, immersion in concentrated sugar solutions, and extensive dehydration to enhance the survival after cryopreservation in plant cells and tissues were reported (Shibli et al. 1999; Tahtamouni and Shibli 1999; Shibli 2000). The result was an increase of the cytoplasmic microviscosity, slowing the water reorganization process required for ice nucleation. Viscosity, which also increases steeply as the system temperature decreases, can reach such high values that molecular movement is virtually stopped. In this situation, named glassy or vitreous state, most chemical reactions and physical processes are considered to be detained, and ice formation is deemed as impossible (Sakai and Engelmann 2007).
Cryopreservation techniques include vitrification (Moukadiri et al. 1999), encapsulation–vitrification (Hirai and Sakai 1999), and encapsulation–dehydration (González-Arnao et al. 1996, 1999; Sakai 2000; Shibli et al. 1999; Shibli 2000). Among these methods, encapsulation–dehydration is widely used because it is applicable to many species (Shibli 2000). Encapsulation–dehydration cryopreservation methods, used with a variety of plant germplasm (Rao et al. 1998), are based on the successive osmotic and evaporative dehydration of plant cells, previous to the LN cooling step (Swan et al. 1999). Dehydration techniques allow more flexibility when handling large sample numbers because the processing is less time-critical than the vitrification techniques sensu stricto (Sakai 2000). Encapsulation–dehydration also avoids the use of toxic cryoprotectants as compared to other methods (Shibli et al. 2006; Rabba’a et al. 2012; Feng et al. 2013).
Differential scanning calorimetry (DSC) has been frequently employed to study ice freezing and vitrification (Rajasekaran 1996; Ashmore 1997). Calorimetric data include information on the amount of frozen and unfrozen water, as well as a clear proof of vitrification and its corresponding glass transition temperature, T G. Thermal analysis of samples during pre-treatment prior to cryopreservation in LN has only been employed previously to examine the processes occurring in alginate-encapsulated Ribes nigrum shoot tips (Benson et al. 1996). The larger volume of alginate relative to the encapsulated sample, together with the physical changes and processes within the bead itself during pre-treatment and cryopreservation, may exert a significant influence on the success of the cryopreservation protocol, and hence on the survival of plant samples.
In spite of the recognized importance of performing both cooling and warming steps extremely fast for the success of the cryopreservation process (Wesley-Smith et al. 2001), there have been no previous reports of the measurement of cooling and warming rates for alginate-encapsulated samples. The more similar data available refer to other plant cryopreservation protocols (Kaczmarczyk et al. 2011; Teixeira, 2013).
Low temperature scanning electron microscopy (cryo-SEM) allows observation of microstructural features with no alteration due to preparation procedures, such as fixation and dyeing (Craig and Beaton 1996). Intra- and extra-cellular ice can be observed, after fracture and etching, and be differentiated from vitrified tissues.
The aim of the experiments was to study the effects of different dehydration times of alginated mint shoot tips on water content, vitrification and their post cryopreservation viability (survival and re-growth). Water content and the vitrification process were studied using thermal analysis techniques (DSC) and cryo-SEM.
Materials and methods
Plant material and shoot tip extraction
Plant material was prepared as described elsewhere (Teixeira et al. 2013). Shoot tips were extracted from in vitro shoots of Mentha × piperita (accession “MEN 186” obtained from the IPK Genebank, Gatersleben, Germany). In vitro plants were monthly subcultured on medium MS (Murashige and Skoog 1962) with 3 % sucrose, and incubated at constant temperature (25 °C) with a photoperiod of 16 h, and a photosynthetic photon flux density (PPFD) of 50 µmol m−2 s−1 from fluorescent tubes. One-node segments were obtained from these shoots, transferred to fresh medium and incubated at alternating temperatures of 25 °C (day) and −1 °C (dark), always with 16 h photo- and thermo-period, 50 µmol m−2 s−1 PPFD, provided by fluorescent tubes. After 3-weeks of culture under these conditions, shoot tips (1–2 mm) were excised from axillary buds.
Shoot tip cryopreservation procedure
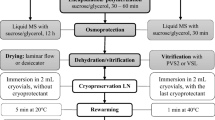
Excised shoot tips were precultured overnight in liquid MS medium (10 tips/2 mL) containing 0.3 M sucrose at 25 °C, over filter paper (Fig. 1). Thereafter, the explants were placed in 3 % (w/v) low viscosity alginic acid (Sigma, USA) in liquid MS without calcium, at pH 5.7. Beads were formed by suspending shoot tips in this solution and dripping them, with the help of a Pasteur pipette, into a calcium chloride solution (MS medium with 100 mM CaCl2 and 0.4 M sucrose). Beads were allowed to polymerize for 30 min. Encapsulated shoot tips were cultured overnight in liquid MS medium with 0.75 M sucrose on a rotary shaker (130 rpm). Beads were blotted dry on sterile filter paper, transferred to an open glass Petri dish and dehydrated for 1, 2, 3, 4, 5 and 6 h in a laminar flow hood. Dehydrated beads (30 per treatment) were placed in 1.0 mL cryovial (10 beads per cryovial) and plunged into LN (Fig. 1). The cryovials were rewarmed in a water bath at 45 °C for 1 min and then at 25 °C for one more minute (Uchendu and Reed 2008). Subsequently, beads were placed on the re-growth medium (MS, with 3 % sucrose and 0.5 mg L−1 6-benzylaminopurine), incubated in the dark for 24 h and thereafter under 16 h photoperiod, at 25 °C. For control samples (–LN), all steps were carried out except immersion in LN and re-warming.

Scheme of the steps of the encapsulation–dehydration protocol employed, see text for more details on media and solutions used
Survival and re-growth were calculated as percentages over the total number of beads used, 4 weeks after culture. Survival was defined including all forms of visible viability (evidence of green structures or callus). Re-growth was defined as the formation of small plantlets. Three replicates (of 10 beads each) were used per treatment. Data were subjected to arcsine transformation prior to analysis of variance and Duncan’s Multiple Range Test (α = 0.05) was used for comparison of means.
Water content determination
Four sets of three specimens were used for determining the dry mater content (dm) of either shoot tips (control and after 0.3 M sucrose incubation step) or beads after the different drying times. It resulted from weighing after oven-drying (~85 °C, 72 h). The water content (W c) was calculated as the difference between the total mass and dm. It was expressed as relative to the fresh weight mass (W c(s)) or the dry mass (W c(dm)).
Differential scanning calorimetry
DSC experiments were performed with a Mettler-Toledo DSC 30 instrument (Mettler-Toledo, Griefsen, Switzerland). Cooling was performed using the calorimeter control (10 °C min−1), or for higher rates, by quenching the samples in LN (see below). The response of beads after each step of the cryopreservation protocol was evaluated. Three beads were included in a DSC pan, which was sealed and weighed. Samples were submitted to a cooling scan from room temperature to −150 °C and a short 5 min equilibration at this temperature, followed by a warming scan (at 10 °C min−1), back to room temperature. Calorimetric data were collected from two replicates per treatment. For study of the quenching method, beads, sealed into a DSC pan, were quickly plunged into LN. Pans were subsequently cold-loaded into a pre-chilled DSC oven (−150 °C) for analysis. Later, pans were punctured and dried in an oven at 85 °C, for 72 h. Thermograms were analysed using the standard procedures provided in the Mettler-Toledo STARe software. Ice thawing thermal events (more precise than freezing events), were used for determination of T f. The routine produced the temperature corresponding to the event onset, T f(onset) (corresponding to the equilibrium freezing temperature) and the peak temperature, T f(peak).
The melting enthalpy ∆H f, proportional to the event area, was also calculated. It was expressed as relative to either the total sample mass (∆H f(s)), its dry mass (∆H f(dm)) or its water content ∆H f(w), to allow easier data comparison. Water fractions: frozen water (W f) and unfrozen water (W u), were calculated by comparison of ∆H f and the pure water freezing enthalpy (∆H *f(w) = 333.4 J g−1 at 0 °C), together with the total water contents previously determined, according to Eq. 1:
Specific enthalpy values (per sample gram) were always used, after the corresponding oven-dry pan weights. Enthalpy values measured at temperatures different from the corresponding phase change equilibrium temperature, 0 °C, are underevaluated, as a result of the difference between water and ice heat capacities (Cp w and Cp i , respectively). So, the value of ∆H *f(w) was corrected after Eq. 2, before applying Eq. 1. Single freezing temperatures, T f(peak), were used, and Cp w and Cp i data were obtained from a calculation routine produced in this laboratory (Otero et al. 2002), after interpolation when required, and other sources (Angell et al. 1973, 1982).
Frozen (W f) and unfrozen (W u) water contents were expressed as relative to the total sample mass (W f(s),W u(s)), its dry mass (W f(dm), W u(dm)) or its total water content (W f(w), W u(w)).
Temperature measurement and cooling and warming rates
Thin T-type thermocouples (copper-constantan) (TC Medida y Control, S.A., Madrid, Spain), with good performance at LN temperatures, were employed to measure cooling and warming rates, using a fast data acquisition equipment MW100-UNV-H04 (Yokogawa, Tokyo, Japan), with a frequency of 20 data per second, basically as described previously (Teixeira 2013). LN and ice were used as controls (−195.8 and 0.2 °C, respectively), calibration being performed frequently between measurements. Cooling and warming rates were defined as the ratio of a predetermined temperature interval T 1–T 2, over the time elapsed between the reported measurement of T 1 and T 2. The determination of cooling and warming rates was carried out, at least in triplicate, during the cooling and warming phases of the described protocols. Thermocouples were calibrated as a part of the measurement routine.
A cartoon of the experimental set employed for cooling and warming rate determination is shown in Fig. 2. Cryovials, containing 10 alginate beads, were handled by means of the attached thermocouple wire, with isolated tongs, to reduce heat losses. For cooling rate determination, cryovials were tempered at 1–4 °C before immersion in LN, and data collection took place from 0 °C till near −196 °C. For warming, cryovials were immersed and stirred in a 40 °C water bath (for 1 min) followed by immersion and stirring in another bath at 25 °C (for 1 min). Measuring took place from the exit from LN till room temperature equilibrium. Rates were calculated after adopting a suitable ice-risk temperature window (cooling: 0 to −150 °C, warming: −150 to 0 °C), by dividing the thermal difference between the window extremes by their time difference.

Experimental setup for thermal change rate measurement for encapsulation–dehydration
Low temperature scanning electron microscopy
Low temperature scanning electron microscopy (cryo-SEM) observations were performed, basically, as previously described (Teixeira et al. 2013), with a Zeiss DSN 960 scanning microscope equipped with a Cryotrans CT-1500 cold plate (Oxford, UK). Cryo-SEM allows sample observations without the need of prior chemical fixing or drying processes.
Three shoot tips or beads containing tips in the same stage of the cryopreservation protocol were fitted on a special bracket with their axes vertically aligned (a cartoon of the tip-containing beads, before placing inside the microscope and after fracture can be seen in Fig. 3), and this piece was immersed into LN under low pressure, physically fixing tissues for microscopic observation. This cooling step was considered equivalent to the cooling process taking place in the actual cryopreservation process, carried out by LN immersion. The holder was, then, placed in the pre-chamber of the Cryotrans cold plate, at −180 °C, and specimens were fractured perpendicularly to their axes, to obtain a suitable observation surface. Later, samples were inserted in the microscope and etching (partial ice sublimation, induced to provide contrast) was performed for 3 min at −90 °C. Once etched and re-cooled, the samples were retrieved into the cryo-preparation chamber and coated with high purity Au, which acts as a conductive contact for electrical charge. Finally, the etched and coated samples were reinstated into the cooled SEM stage and observed under secondary electron mode, working at −150/−160 °C, using an accelerating voltage of 15 kV.

Cartoon showing the three mint tips-containing beads on the microscopy holder before insertion in the microscope (a) and after equatorial freeze fracture (b)
Results and discussion
Water content reduction during the drying period
The dry mass (dm) and water content of shoot tips (control and 0.3 M sucrose precultured tips) and beads containing shoot tips (after different drying times: 0–6 h), derived from weight-difference after extensive drying, are shown in Table 1. Water content was expressed in relation to the total sample mass (W c(s)) and the dry mass (W c(dm)). The water content of tips slightly decreased with the preculture in medium with 0.3 M sucrose.
The initial water content of beads was lower than that of precultured tips, as a result of the culture of beads in 0.75 M sucrose. For increasing drying times water content decreased asymptotically, with the highest water reduction taking place between 1 and 3 h of drying time. Longer drying periods were associated to slight or non-significant water content variations, when compared with the experimental error. An associated bead size decrease and hardening could also be observed.
Calorimetric studies on water status
Calorimetric measurements were carried out for shoot tips (control and precultured) and tip-containing beads for 0–6 h drying times. Figure 4 shows typical scans obtained for beads at the different drying times. Inserts help visualizing the small glass transitions, when present. The parameters derived from DSC data analysis (melting exotherm temperatures and enthalpies and the derived frozen water contents) are shown in Table 1. Clearly, samples could be divided in two sets, corresponding to higher and lower water contents. Experiments with non-alginated tips and beads desiccated for 0, 1 or 2 h presented a melting exotherm, implying that part of the water in these systems became frozen upon cooling. Meanwhile, samples at drying times 3–6 h showed no trace of melting events, indicating no water was frozen. Instead, these samples exhibited glass transitions, proving these beads became vitrified upon cooling.

Typical DSC thermograms corresponding to re-warming processes of shoot tips with beads of the encapsulation–dehydration protocol. Beads were desiccated under the air flow of a laminar flow bench for different times: T0 = 0 h (no desiccation); T1 = 1 h; T2 = 2 h; T3 = 3 h; T4 = 4 h; T5 = 5 h and T6 = 6 h. Each experiment was performed with three beads. The insets show an expanded thermogram section which allows seeing the glass transition. Scanning rate was 10 °C min−1. The ordinates scale of the thermograms baseline is arbitrary
Data analysis (Table 1) allowed drawing a perfect parallelism between water content and the calorimetric behaviour. The temperatures of the melting process (when present) decreased as water content did, showing that the solute concentration increased. The area of the curves, i.e., the melting enthalpy, also decreased with water content, as well as the derived frozen water content. While W f(w) for control tips was approx. 80 %, this amount decreased to nearly 50 % in beads, after 2 h desiccation, which was the longer drying time showing freezing processes. This, added to the approx. 40 % reduction in total water content at T2, implies that the mass of frozen water was only 20 % of the sample total mass. The ratio of frozen to unfrozen water decreased in control and precultured tips and with increasing drying times, and reached a value of 1 at the highest drying time showing a freezing event, i.e., at 2 h desiccation (Fig. 5).

Ratio W f(dm) W −1u(dm) for mint shoot tips (control and precultured on 0.3 M sucrose) and shoot tip-containing beads (for drying times 0–6 h). Bars standard error
Glass transition temperatures (Table 1) increased with the progress of the drying process. The increase was observed in 3–5 h desiccation time, while there was no difference from 5 to 6 h, which corresponded to the very small or constant water content reduction at these times.
Beads and tips studied by DSC in a similar way after LN quench cooling presented a very similar behaviour (data not shown), but their freezing events were displaced to lower temperatures. Beads, dried from 3 to 6 h, showed no freezing processes and the glass transitions observed in these cases were unchanged, within experimental error.
Cooling and warming rate determination
In this work, an ice-risk formation window of 0 to −150 °C was considered for cooling and warming rate calculation, instead of the whole interval from 0 to −196 °C. This window comprises of the regions were ice formation can take place, though with a very low probability or in a very long time (i.e., in very slow cooling or warming processes). The selected window is widely over-dimensioned compared to the actual ice formation risk temperature interval for cryopreserved systems, as its equilibrium freezing temperature is always well under 0 °C, due to its high solute and low water contents. In contrast, the glass transition temperature, under which the possible ice formation area ends, although highly variable, tends to be located well over the −150 °C limit. Other narrower windows would yield higher rates, while considering the duration of the process all the way down to LN temperature (−196 °C) would yield meaningless and exceedingly long times (Teixeira et al. 2013).
Cooling (Fig. 6) and warming (Fig. 7) processes followed an approximately symmetrical behaviour with only small differences among samples. It must be noted that natural differences in samples and manipulation variations during normal cryopreservation procedures contribute to a higher dispersion in data (Teixeira 2013).

Temperature evolution, during cooling by immersion in LN, of cryovials with 10 alginate beads containing mint shoot tip and treated after the encapsulation–dehydration protocol. Beads were desiccated under the air flow of a laminar flow bench for different times: T0 = 0 h (no desiccation); T1 = 1 h; T2 = 2 h; T3 = 3 h; T4 = 4 h; T5 = 5 h; T6 = 6 h; and Control = cryovial without beads. Data are the mean of at least three repeats

Temperature evolution, during warming in a water bath at 45 ºC for 1 min and then at 25 °C for 1 min of cryovials with 10 alginate beads containing mint shoot tip and treated after the encapsulation–dehydration protocol. Beads were desiccated under the air flow of a laminar flow bench for different times: T0 = 0 h (no desiccation); T1 = 1 h; T2 = 2 h; T3 = 3 h; T4 = 4 h; T5 = 5 h; T6 = 6 h; and Control = cryovial without beads. Data are the mean of at least three repeats
The cooling rates obtained with beads dried for different times (Table 2) showed an increase easy to be corelated to the water content decrease (Table 1). Rates could be related to the samples relevant physical characteristics: mass, heat capacity and thermal conductivity. Besides the dependence on mass, larger water contents imply larger global heat capacities. In this case, both sample mass and its heat capacity decreased with drying time. Heat conductivity also plays a role in determining cooling and warming rates. Factors such as the position of beads in the cryovials and the contact between the wall and beads are also important. These thermal contacts are favoured by the higher water content in beads.
The larger variations in rates were found between 2 and 3 h drying time, and at longer periods the variation was quite small, also correlating with the asymptotic water content reduction. Warming rates behaved in a similar way, although the observed warming rates were smaller than the cooling ones.
Literature shows very few experimental measurements of these rates, in spite of its generally assumed importance. Only Hirai (2011) reports a value of −2.1 °C min−1 for encapsulated samples, although his plant system was different and the comparability is poor, as this author followed an encapsulation–vitrification protocol. The rates reported here are higher by nearly two magnitude orders. Additionally to the noted experimental differences, this large difference could be due to the rate calculation procedure followed in this work, were only the temperature change between 0 and −150 °C was considered.
The warming process was performed after the encapsulation–dehydration protocol, and included a 1 min warming in a 45 °C water bath plus 1 min in a 25 °C bath. For beads with the highest water content, the time was insufficient to reach temperatures over 0 °C (Table 2). To complete the warming process at room temperature would lengthen the time under 0 °C and could increase the risk of ice formation.
Low temperature scanning electron microscopy
The microstructure of alginate gel beads exposed to the extreme conditions of the drying process (6 h) was observed by cryo-SEM, and compared to that of non-desiccated ones. Observation with secondary and backscattered electrons was compared, and although the same basic information was obtained with both, cellular structures appeared clearer with backscattered electrons, which were used in the micrographs presented.
Figure 8 shows pictures corresponding to these conditions (sections not including shoot tips). The water content reduction from a–c to b–d beads implied a significant increase of solute concentration in the remaining water. Although not directly measured, it could be roughly estimated, considering an initial 0.75 M sucrose concentration in the T0 beads, which, for an approximate tenfold decrease in W c(dm) between T0 and T6, would give a final 7.5 M sucrose. This would give rise to a strong reduction in molecular mobility for high drying times, which caused the system to vitrify when quickly cooled at LN temperatures (in the microscope stage).

Cryo-SEM micrographs of alginate beads. View of beads external surface at drying time 0 (a) and 6 h (b), and internal cryofracture surface, at drying time 0 (c) and 6 h (d). The bars correspond to 10 µm
The etching procedure employed for creating contrast in cryo-SEM removes part of the ice formed in the cooling of the sample in the microscope (always small crystals, due to the high cooling rate and the small size of the sample) by sublimation, leaving darker “void” regions separated by cleared “ridges” of freeze-concentrated solution. Glassy state has a near null sublimation rate, so vitrified solutions do not present enhanced contrast after the etching process (Angell et al. 1982; Umrath 1983). This was considered a suitable method for studying vitrified tissues and their microstructure and was employed in previous works of this group (Teixeira et al. 2012, 2013).
Figure 9 shows cryo-SEM micrographs corresponding to mint shoot tips at different stages of the encapsulation–dehydration protocol. a–g correspond to the progressive drying times. Pictures a–d show microstructural intracellular details resulting from the etching ice sublimation procedure, and in the conditions of microscopic observation that are assumed to be equivalent to those of the rapid cooling in LN used in normal cryopreservation protocols, intracellular ice was present and hence, damage to cell function occurred. On the other hand, pictures e–g, corresponding to the longer drying times, show no structural details, implying that water could not sublimate during etching and therefore that these tissues were in glassy state.

Cryo-SEM micrographs of mint shoot tips in different stages of the encapsulation–dehydration protocol. Drying times (hours): a (0), b (1), c (2), d (3), e (4), f (5), g (6). h Tip growing for 1 week, after warming. The bars correspond to 10 μm
The four first pictures (a–d) show intracellular ice crystals of decreasing size as the cytoplasm concentration increased with water reduction. Apices after 1 and 2 h desiccation (T1 and T2; b and c) exhibited extracellular solutions partially vitrified that could be also associated to the higher sucrose concentration.
Meanwhile, cells at T3 (Fig. 9 d) show a mix of intracellular ice with vitrified regions. The reduction of ice crystals size is a direct effect of the increase in viscosity, allowing more ice nuclei to grow instead of favouring the growth of only a few crystals.
The cells shown in Fig. 9 h correspond to a cryopreserved and recovered specimen, after 1 week of successful culturing. Its water and solute contents would be similar to that observed in material at the starting point of the process (as reported by Teixeira et al. 2012), and ice crystals, larger than in Fig. 9 a, could be observed, what corresponds to a higher water content.
Cryopreserved shoot tips survival and re-growth
Survival and re-growth after 4 weeks culture on recovery medium were evaluated for shoot tips cryopreserved following the encapsulation–dehydration protocol with different air-dehydration times and subsequent cooling in LN (Fig. 10). In most cases viability and re-growth rates were fairly similar within treatment. Control specimens, not immersed in LN, had nearly a 100 % survival, proving the good initial performance of shoot tips.

Mint shoot tip survival and re-growth percentages after dehydration for different times following the encapsulation–dehydration protocol. Observations were made after a 4-week recovery period. Beads were desiccated under the air flow of a laminar flow bench for different times: T0 = 0 h (no desiccation); T1 = 1 h; T2 = 2 h; T3 = 3 h; T4 = 4 h; T5 = 5 h; T6 = 6 h; and Control = before cooling in LN. Data are the mean of at least three repeats. Means of survival or re-growth with the same letter are not significantly different according to the Duncan’s Multiple Range Test at α = 0.05. Bars standard error
The T0 and T1 high water content samples showed complete lack of survival or re-growth. Intermediate water contents (T2 and T3) had rates of less than 50 % for both these parameters, while the dryer samples (T4, T5 and T6) showed high and similar survival and re-growth. The re-growth percentage for alginated shoot tips exposed to 6 h of dehydration was 93.3 %.
General discussion
The general behaviour of mint shoot tips following the encapsulation–dehydration cryopreservation protocol at different air-drying times was clear and consistent, in spite of the inherent variability of plant samples and of the whole process. Water content in beads (and hence in the encapsulated tips) decreased with drying time, reaching at T3 approximately stable values (W c(s) ~ 0.25 gwater g −1sample , W c(dm) ~ 0.30 gwater g −1dm ). These water contents were in the range of critical values reported for encapsulated shoot tips (Dereuddre et al. 1990; Uragami et al. 1989). Damages associated with water content of these orders were reported for different plant germplasm species (González-Benito et al. 1998, 2004; Sun 1999).
Most of the other parameters and observations behaved in parallel to water content. Both cooling and warming rates increased as the drying time did (Figs. 6, 7). Fast cooling and warming processes reduce the probability of ice formation and increase that of vitrification. Nevertheless, the increase in these rates for the different drying times is relatively small and quite likely not the main source of the different success of the cryopreservation process.
A water content reduction would mean an increased solute concentration in the remaining water. This results, for both bead and shoot tip, in higher viscosity (reduced molecular mobility) and hence, an increased probability of vitrification as they get quickly cooled down. Low temperature scanning electron microscopy showed (Figs. 8, 9) ice crystallization in samples with higher water content but after 4 h drying, vitrification took place and ice crystals were not visible (with the T3 samples in an intermediate situation).
The water freezing parameters obtained from DSC (Table 1) agreed with the previous observations by showing melting processes fully compatible with increasing solute concentrations (decrease of freezing temperature and reduction of frozen water content) up to 3 h of desiccation when no ice formation could be detected. The calorimetric glass transitions that were visible after this drying time (3 h) confirmed the cryo-SEM observations; glass transition temperature increased up to 5 h desiccation, in accordance with the progress of the dehydration process.
The observed viability of cryopreserved specimens (survival and recovery, as a global physiological evaluation of the success of the procedure), is in good agreement with the results reported for this protocol (Uchendu and Reed 2008) and confirmed the thermodynamic results and the images observed.
The encapsulation–dehydration procedure submits specimens to prolonged dehydration and high sucrose concentration stresses. This may cause damage in the recovered plants, such as genetic alterations (Martín et al. 2011), but such damages were not evaluated in the viability analysis of this experiment. This study showed that the asymptotic reduction of water content and the associated variation of most parameters make it unnecessary to employ long desiccation times to protect samples from ice formation. DSC observations implied that 3 h drying could be enough to avoid the formation of ice (even operating at a slow cooling rate: 10 °C min−1). Nevertheless, cryo-SEM showed how the shoot tips cytoplasm at 3 h drying time might be not completely ice-free, as some crystal presence was observed. This was confirmed by the viability data, in which samples desiccated for 3 h showed an intermediate response, implying that for some of the specimens, lethal ice formation may have taken place.
A clearer picture was made by the glass transition temperature evolution in the lower water content samples. Although water content data indicated no further changes from 3 h desiccation onwards, there was a distinct increase of T G from 3 to 5 h desiccation, where a stable situation was reached.
In conclusion, the success of the encapsulation–dehydration procedure, a practical and efficient method for cryopreservation of shoot tips, can be associated to cytoplasmic vitrification and avoidance of intracellular ice formation. The combined results of DSC and cryo-SEM are in good agreement and their joint use allows the understanding of the physical processes responsible for the variability of the cryopreservation process.
References
Angell CA, Shuppert J, Tucker JC (1973) Anomalous properties of supercooled water. Heat capacity, expansivity, and proton magnetic resonance chemical shift from 0 to −38°. J Phys Chem 77:3892–3899
Angell CA, Oguni M, Sichina WJ (1982) Heat capacity of water at extremes of supercooling and superheating. J Phys Chem 86:998–1002
Ashmore SE (1997) Status report on the development and application of in vitro techniques for the conservation and use of plant genetic resources. International Plant Genetic Resources Institute, Rome
Benson EE, Reed BM, Brennan RM, Clacher KA, Ross DA (1996) Use of thermal analysis in the evaluation of cryopreservation protocols for Ribes nigrum L. germplasm. CryoLetters 17:347–362
Craig S, Beaton CD (1996) A simple cryo-SEM method for delicate plant tissues. J Microsc 182:102–105
Dereuddre J, Scottez C, Arnaud Y, Duron M (1990) Résistance d’apex caulinaires de vitroplants de Poirier (Pyrus communis L. cv Beurré Hardy), enrobés dans l’alginate, à une déshydratation puis à une congélation dans l’azote liquide: effet d’un endurcissement préalable au froid. C R Acad Sci 10:317–323
Engelmann F (1997) In vitro conservation methods. In: Callow JA, Ford-Lloyd BV, Newbury HJ (eds) Biotechnology and plant genetic resources: conservation and use. CAB International, Wellingford, pp 119–162
Engelmann F, Takagi H (2000) Cryopreservation of tropical plant germplasm—current research progress and applications. Tsukuba & International Plant Genetic Resources Institute, Rome
Feng CH, Cui ZH, Li BQ, Chen L, Ma YL, Zhao YH, Wang QC (2013) Duration of sucrose preculture is critical for shoot regrowth of in vitro-grown apple shoot-tips cryopreserved by encapsulation–dehydration. Plant Cell Tissue Organ 112:369–378
González-Arnao MT, Moreira T, Urra C (1996) Importance of pre growth with sucrose and vitrification for the cryopreservation of sugarcane apices using encapsulation–dehydration. CryoLetters 17:141–148
González-Arnao MT, Urra C, Engelmann F, Ortiz R, Fe C (1999) Cryopreservation of encapsulated sugarcane apices: effect of storage temperature and storage duration. CryoLetters 20:347–352
González-Benito ME, Núñez-Moreno Y, Martín C (1998) A protocol to cryopreserve nodal explants of Antirrhium microphyllum by encapsulation–dehydratation. CryoLetters 19:225–230
González-Benito ME, Clavero-Ramírez I, López-Aranda JM (2004) Review. The use of cryopreservation for germoplasm conservation of vegetatively propagated crops. Span J Agric Res 2:341–351
Hirai D (2011) Gelled droplet vitrification improves recovery of cryopreserved potato germplasm. CryoLetters 32:287–296
Hirai D, Sakai A (1999) Cryopreservation of in vitro-grown axillary shoot-tip meristems of mint (Mentha spicata L) by encapsulation vitrification. Plant Cell Rep 19:150–155
Kaczmarczyk A, Rokka VM, Keller ERJ (2011) Potato shoot tip cryopreservation. A review. Potato Res 54:45–79
Martín C, Cervera MT, González-Benito ME (2011) Genetic stability analysis of chrysanthemum (Chrysanthemum x morifolium Ramat) after different stages of an encapsulation–dehydration cryopreservation protocol. J Plant Physiol 168:158–166
Matsumoto T, Saki A, Nako Y (1998) A novel preculturing for enhancing the survival of in vitro-grown meristems of wasabi (Wasabia japonica) cooled to −196°C by vitrification. CryoLetters 19:27–36
Moukadiri O, Deming J, O’Connor JE, Cornejo MJ (1999) Phenotypic characterization of the progenies of rice plants derived from cryopreserved calli. Plant Cell Rep 18:625–632
Murashige T, Skoog F (1962) A revised medium for rapid bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Otero L, Molina-García AD, Sanz PD (2002) Some interrelated thermophysical properties of liquid water and ice. I. A user-friendly modelling review for food high-pressure processing. Crit Rev Food Sci Nutr 42:339–352
Rabba’a MM, Shibli RA, Shatnawi MA (2012) Cryopreservation of Teucrium polium L. shoot-tips by vitrification and encapsulation–dehydration. Plant Cell Tissue Organ 110:371–382
Rajasekaran K (1996) Regeneration of plants from cryopreserved embryogenic cell suspension and callus cultures of cotton (Gossypium hirsutum L.). Plant Cell Rep 15:859–864
Rao PS, Suprasanna P, Ganapathi TR, Bapat VA (1998) Synthetic seeds: concepts, methods and application. In: Srivastava PV (ed) Plant tissue culture and molecular biology. Narosa Publishing House, New Delhi, pp 607–619
Sakai A (2000) Development of cryopreservation techniques. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. International Plant Genetic Resources Institute, Rome, pp 1–7
Sakai A, Engelmann F (2007) Vitrification, encapsulation–vitrification and droplet-vitrification: a review. CryoLett 28:151–172
Shibli RA (2000) Cryopreservation of black iris (Iris nigricans) somatic embryos by encapsulation–dehydration. CryoLetters 21:39–46
Shibli RA, Smith MAL, Shatnawi M (1999) Pigment recovery from encapsulated-dehydrated Vaccinium pahalae cryopreserved cells. Plant Cell Tissue Organ 55:119–123
Shibli RA, Shatnawi MA, Subaih WS, Ajlouni MM (2006) In vitro conservation and cryopreservation of plant genetic resources: a review. World J Agric Sci 2:372–382
Sun WQ (1999) State and phase transition behaviors of Quercus rubra seed axes and cotyledonary tissues: relevance to the desiccation sensitivity and cryopreservation of recalcitrant seeds. Cryobiology 38:372–385
Swan TW, O’Hare D, Gill RA, Lynch PT (1999) Influence of preculture conditions on the post-thaw recovery of suspension cultures of Jerusalem artichoke (Helianthus tuberosus L.). CryoLetters 20:325–336
Tahtamouni RW, Shibli RA (1999) Preservation at low temperature and cryopreservation in wild pear (Pyrus syriaca). Adv Hortic Sci 13:156–160
Teixeira AS (2013) Thermodynamic studies of vitrification processes in plant germplasm cryopreservation. Ph.D. thesis. Universidad Politecnica de Madrid, Madrid, Spain
Teixeira AS, González-Benito ME, Molina-García AD (2012) Tissue and cytoplasm vitrification in cryopreservation monitored by low temperature scanning electron microscopy (cryo-SEM). In: Méndez-Vilas A (ed) Current microscopy contributions to advances in science and technology. Formatex, Badajoz, pp 872–879
Teixeira AS, González-Benito ME, Molina-García AD (2013) Glassy state and cryopreservation of mint shoot tips. Biotechnol Prog 29:707–717
Towill LE (1996) Vitrification as a method to cryopreserve shoot tips. In: Trigiano RS, Gray DJ (eds) Plant tissue culture concepts and laboratory exercises. CRC Press, Boca Raton, pp 297–304
Towill LE (2002) Cryopreservation of Mentha (mint). In: Towill LE, Bajaj YPS (eds) Cryopreservation of plant germplasm II. Springer, Heidelberg, pp 151–163
Uchendu EE, Reed BM (2008) A comparative study of three cryopreservation protocols for effective storage of in vitro-grown mint (Mentha spp.). CryoLetters 29:181–188
Umrath W (1983) Calculation of the freeze-drying time for electron-microscopical preparations. Mikroskopie 40:9–34
Uragami A, Sakai A, Nagai M, Takahashi T (1989) Survival of cultured cells and somatic embryos of Asparagus officinalis cryopreserved by vitrification. Plant Cell Rep 8:418–421
Wesley-Smith J, Walters C, Pammenter NW, Berjak P (2001) Interactions among water content, rapid (nonequilibrium) cooling to −196 °C, and survival of embryonic axes of Aesculus hippocastanum L. seeds. CryoLetters 42:196–206
Acknowledgments
This work has been carried out thanks to project “CRYODYMINT” (AGL2010-21989-C02-02) of the Spanish Ministry of Science and Innovation. A.S. Teixeira was supported by the CSIC, within the JAE-Pre program, partially funded from the European Social Fund. It is also acknowledged the expert microscopic work of F. Pinto, who sadly passed away, shortly after this work was completed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Teixeira, A.S., González-Benito, M.E. & Molina-García, A.D. Determination of glassy state by cryo-SEM and DSC in cryopreservation of mint shoot tips by encapsulation–dehydration. Plant Cell Tiss Organ Cult 119, 269–280 (2014). https://doi.org/10.1007/s11240-014-0531-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0531-3