
Abstract
A protocol for plant regeneration from protoplasts of Musa acuminata cv. Mas (AA) via somatic embryogenesis was developed. Viable protoplasts were isolated from embryogenic cell suspensions at a yield of 1.2 × 107 protoplasts/ml packed cell volume (PCV). Liquid and feeder layer culture systems with medium-A and medium-B were used for protoplast culture. In liquid culture system, medium-B was more efficient for inducing cell division (17.5% at 14 days) and colony formation (6.7% at 28 days) than medium-A. However, all protoplast-derived cell colonies (PDCC) obtained from liquid culture system could not develop further. In feeder layer culture system, there was no significant difference between medium-A and medium-B on cell division and colony formation of the cultured protoplasts, and the cell division frequency at 14 days and colony formation frequency at 28 days were 24.5% and 11.2%, respectively, in medium-B. Comparative study on the effects of BAP (2.2 μM, 4.4 μM, 8.8 μM), zeatin (0.4 μM, 0.8 μM, 1.2 μM) and TDZ (0.2 μM, 0.4 μM, 0.6 μM) on embryo formation of PDCC from feeder-layer culture indicated that TDZ was best. TDZ at 0.4 μM induced 7906 mature embryos per ml PCV PDCC, which was 4-fold the frequency as with BAP at 4.4 μM, 7.5-fold as with zeatin at 0.8 μM and 150-fold as control medium (no mentioned cytokinins) after 45 days on M3 medium. About 44% of the mature embryos were converted into plantlets with poor root system after subculture on M4 medium. Root further development of regenerated plantlets was promoted by addition of activated charcoal (AC) to MS basal medium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Banana (Musa spp.) is the most important fruit crop in tropical and subtropical countries. The plants are currently being threatened by the spread of viral and fungal disease, which cause a significant reduction in yield and quality. Classical breeding methods have limited success due to the high sterility and triploidy of most cultivated bananas. Somatic hybridization through protoplast fusion to improve disease resistance has been applied with several crops, e.g. Solanum tuberosum L. (Fock et al. 2000) and Oryza sativa L. ssp. japonica (Yan et al. 2004), and could be used to develop new varieties of banana (Matsumoto et al. 2002; Assani et al. 2005). A number of studies on banana protoplast culture have been reported since Bakry (1984) reported the first successful isolation of viable banana protoplasts. It has been shown that besides cultivars the successful protoplast culture of banana and plantain would be dependent on cultural technologies such as application of feeder-layer (Panis et al. 1993; Assani et al. 2001), and the preferred donor material for protoplast isolation has been shown to be embryogenic cell suspensions (ECS) (Assani et al. 2002). The effects of factors such as protoplast isolation, plating density, culture method, nurse cells, medium composition etc. on cell division and microcolony formation, have already been evaluated by Panis et al. (1993) and Assani et al. (2001, 2006). Until now, the cultivars where successful plant regeneration from protoplasts was reported include cooking banana Bluggoe (Musa spp., ABB group) (Megia et al. 1993; Panis et al. 1993), Brazilian dessert banana (Musa spp., AAB group) (Matsumoto and Oka 1998), Grande Naine (Musa spp., Cavendish AAA group) (Assani et al. 2001; Assani et al. 2002), Gros Michel (Musa spp., Cavendish AAA group), Currare Enano and Dominico (Musa spp., Plantain AAB group) (Assani et al. 2002). However, successes with protoplast culture of banana have still been limited and plant regeneration from protoplast is not yet routine or efficient for the majority of banana cultivars. Thus, a study on the establishment of a reproducible regeneration protocol of protoplasts, applicable to various banana varieties, is required.
Musa acuminata cv. Mas (AA) is a popular commercial dessert variety planted in South East Asia. It is favored by consumers for its special sweet taste and thin peel, but this cultivar is highly susceptible to Fusarium oxysporun f. sp.Cubense race 4. It is possible to improve its resistance to this disease through protoplast culture and somatic hybridization. In previous study, we established the ECS of this cultivar from male inflorescence buds (Wei et al. 2005). To our knowledge, there are no reports on protoplast culture of dessert diploid banana though Megia et al. (1992) reported the protoplast sustained divisions leading to callus formation of wild diploid AA banana Musa acuminata ssp. burmannica cv long Tavoy without plantlet regeneration, and Assani et al. (2002, 2005) reported plants regeneration from protoplasts of cooking diploid AA banana SF265, Col49 and IRFA 903. The main objectives of this research were to establish a protocol for plant regeneration from protoplasts of this cultivar, and to study some factors which influence the efficiency of plant regeneration through somatic embryogenesis. In this study we report for the first time that TDZ showed a strong effect on promotion of embryo formation of the protoplast-derived cell colonies (PDCC), and we also showed that activated charcoal (AC) significantly stimulated the root development of regenerated plantlets.
Materials and methods
Plant materials and cultural media
The donor plant, Musa acuminata cv. Mas (AA), a popular commercial dessert variety planted in the South East Asian region, was kindly provided by the Pomology Fruit Research Institute of Guangdong Academy of Agricultural Sciences (China). All media used in this study were listed in Table 1.
Initiation and maintenance of embryogenic cell suspensions
ECS of Musa acuminata cv. Mas (AA), which composed of several cells to more than hundreds cells (Wei et al. 2005), were initiated from male inflorescence buds according to our previous protocol (Wei et al. 2005). Briefly, immature male flowers were excised and cultured on M1 medium (Table 1) (Escalant et al. 1994; Côte et al. 1996) for callus induction. After 5–6 months of culture, friable embryogenic calli were selected and subsequently cultured in liquid M2 medium (Table 1) (Côte et al. 1996) to initiate cell suspensions. After sieving selection of the cultures using a stainless mesh with pore sizes of 154 μm at 15 day-intervals for 3 months, homogeneous and yellow ECS were obtained. The stable ECS routinely used in this study were subcultured every 2 weeks at 1.5% (v/v) dilution in 100-ml Erlenmeyer flasks containing 30 ml of M2 medium on a rotary shaker (100 rpm) at 27°C in the dark.
Protoplast isolation
ECS after 3–5 days of subculture were used for protoplast isolation. The cells were sieved through a 154 μm stainless mesh to select the small cell aggregates. 2 ml packed cell volume (PCV) of this ECS were subsequently incubated in 10 ml protoplast enzyme solution containing 3.5% (w/v) cellulase R-10 (Yakult Pharma., Tokyo, Japan), 1% (w/v) macerozyme R-10 (Kinki Yakult, Mishino., Japan), 0.15% (w/v) pectolyase Y-23 (Seishin Pharma., Japan), 204 mM KCl and 67 mM CaCl2 (pH 5.7). Enzymatic digestion was carried out on a gyratory shaker (30 rpm) at 27°C in the dark for 8–10 h.
Following digestion, the mixture was sieved through a 74/25 μm stainless mesh combination and then collected by centrifugation at 50g for 5 min. The pellet was resuspended in washing solution [204 mM KCl, 67 mM CaCl2, 0.5 mM 2-N-morpholino ethanesulfonic acid (MES) (Sigma) and 10% (w/v) mannitol, pH5.7] and centrifuged twice at 50g for 5 min each. Protoplast viability was determined by fluorescein diacetate (FDA) staining (Widholm 1972). Protoplast yield was estimated using a Nageotte hematocytometer immediately after purification, only viable protoplasts were counted. The presence or absence/regeneration of cell wall was examined after isolation and during culture with Calcofluor White under UV microscope (Nikon) (Nagata and Takebe 1970).
Protoplast culture
Protocol for plant regeneration from protoplast culture was shown in Fig. 1. We used two culture systems (liquid and feeder-layer culture) and two media (medium-A and medium-B) (Table 1) in each culture system to study the optimal protocol for protoplast culture. The pH of the media was adjusted to 5.7 with HCl/NaOH before filter-sterilization.

Protocol for plant regeneration from protoplasts of Musa acuminata cv. Mas (AA) via somatic embryogenesis. The media used here were showed in Table 1
Feeder-layer culture system
Preparation of feeder-layer was based on the protocol reported by Assani et al. (2001) slightly modified. ECS of cv. Mas (AA) were used as nurse cells. The nurse cultures were prepared (1 day before protoplast isolation) as follows: (1) The ECS were initially sieved through a 154 μm stainless mesh in order to select the small cell aggregates. The feeder-layer medium (PCM) (Table 1) was sterilized by filtration. Nurse cell suspensions were mixed with 100 ml of double-concentrated PCM to obtain a final cell concentration of 10% (v/v). (2) Sea-plaque Agarose (1.2 g) (Sigma) was dissolved in 100 ml water and then autoclaved (pH 5.7); when the temperature of agarose solution decreased to 30–35°C, it was gently mixed with 100 ml double-concentrated PCM containing nurse cells. (3) Aliquots of 20 ml of this mixture were poured into Petri dishes (9.5 cm in diameter). After solidification, the medium was covered with a sterilized filter, and 1 ml of the protoplast suspension in medium-A or medium-B at density of 1 × 106 protoplasts/ml was transferred onto the filter. The Petri dishes were sealed with Parafilm.
Liquid culture system
Protoplasts were suspended in 2 ml of liquid “medium-A” or “medium-B” at a density of 1 × 106 protoplasts/ml and then transferred into small Petri dishes (5.5 cm in diameter), which were sealed with Parafilm.
All cultures were maintained at 27°C in the dark. Division frequency of cultured protoplasts was recorded on day 14 after culture and colony formation frequency on day 28.
Initiation of cell suspensions of protoplast-derived cell colonies
After 45 days of culture, cell colonies derived from feeder-layer and liquid culture system in medium-B were collected and resuspended in liquid M2 medium (Table 2). Aliquots of 0.3 ml PCV of those cell colonies were cultured in 50-ml Erlenmeyer flasks containing 10 ml liquid M2 medium. The cultures were kept on a gyratory shaker at 100 rpm at 27°C in the dark.
Somatic embryogenesis
After culture in liquid M2 medium for 1 month, 10% ml PCV of cell suspensions derived from liquid and feeder-layer culture system in medium-B were used for somatic embryogenesis. One milliliter of these cell suspensions was transferred into Petri dishes (9.5 cm in diameter) with 20 ml of M3 medium (Table 1). The M3 medium without cytokinins (control) or supplemented with BAP (2.2 μM, 4.4 μM, 8.8 μM), zeatin (0.4 μM, 0.8 μM, 1.2 μM) or TDZ (0.2 μM, 0.4 μM, 0.6 μM) were used for studying the effects of cytokinins on the somatic embryogenesis. TDZ and zeatin were added by filter sterilization to the autoclaved medium. All cultures were maintained at 27°C in the dark, and subcultured every 15–20 days. The frequency of embryo formation was calculated 45 days after plating of PDCC on M3 medium. The bipolar embryos (mature embryos) were counted under dissecting microscope and the frequency of embryo formation was recorded as number of mature embryos per ml PCV PDCC.
Mature bipolar embryos were picked out from M3 medium with 0.4 μM TDZ and subcultured every 15–20 days on M4 medium (Table 1) for germination until embryos developed into plantlets with shoot of 2–3 cm long. Such regenerated plantlets were transferred onto rooting medium (RM) (Table 1) which consisted of basal MS medium with or without 0.1% AC to promote root further development. All cultures were maintained at 27°C under 16 h photoperiod (65 μmol m−2 s−1). The conversion frequency of embryos into plantlets was calculated 30 days after subculturing of mature embryos on M4 medium and was recorded as the number of plantlets/mature embryos initially incubated × 100%. Rooting responses were recorded 25 days after transferring regenerated plantlets in RM, including the number of roots (roots longer than 1 cm), the average length of roots and height of plantlets.
Statistical analysis
All experiments were repeated at least three times. The data collected was analyzed using Duncan’s multiple range test. Least significant differences were calculated at the 5% level of probability.
Results
Protoplast isolation
Protoplasts were released from ECS after 8–10 h of incubation in enzyme solution. Protoplast yield was 1.2 × 107 protoplasts/ml PCV ECS, and they showed over 85%-90% viability as assessed with FDA staining. The freshly isolated protoplasts were rich in cytoplasm and relatively uniform in size (Fig. 2a). These protoplasts showed no fluorescence after staining with Calcoflour White (Fig. 2a, 2b).

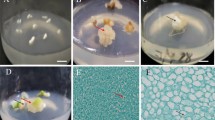
Plant regeneration from protoplasts of Musa acuminata cv. Mas (AA) via somatic embryogenesis (a) Freshly isolated protoplasts, bar 50 μm. (b) Freshly isolated protoplasts stained with Calcofluor White showed no fluorescence, cell wall debris showed fluorescence, bar 50 μm. (c) Arrow 1: dead protoplast; Arrow 2: elongated protoplast after 2 d of culture; bar 50 μm. (d) Arrow 1: dead protoplast stained with Calcofluor White showed no fluorescence; Arrow 2: elongated protoplast stained with Calcofluor White showed fluorescence; bar 50 μm. (e) Protoplast became large and oval in shape (arrow 1) and the first division of the protoplasts (arrow 2) in feeder-layer culture system, bar 50 μm. (f) Non-embryogenic cell colonies derived from liquid culture system 20 d after incubated in medium-B, bar 50 μm. (g) Embryogenic cell colonies derived from feeder-layer culture system 20 d after incubated in medium-B, bar 50 μm. (h) Cultures containing somatic embryos at different development stages, bar 4 mm. (i) Germinating embryos, bar 2 cm. (j) 1: Plantlets regenerated on MS medium with activated charcoal; 2: Plantlets regenerated on MS medium; bar 5 cm. (k) Plantlets regenerated via somatic embryogenesis from protoplasts by feeder-layer culture; bar 4 cm
Protoplast culture
Cell division and colony formation in feeder-layer culture system
Protoplasts cultured on feeder-layer in medium-A and medium-B gave rise to no significant difference with regard to cell division and colony formation (Table 2). Most protoplasts in either media became large and oval in shape 2 days after plating on feeder-layer (Fig. 2c-2, d-2, e-1). Fluorescence of protoplasts within 48 h of culture, using the fluorescent dye Calcofluor White, indicated cell wall regeneration (Fig. 2d-2). First divisions of protoplasts were observed at 5 days (Fig. 2e-2). After 25–30 days of culture, cell colonies were visible by the naked eyes (Fig. 2g). Microscopic observations showed that cells in the colonies were tightly packed and with a dense cytoplasm, suggesting that they were embryogenic (Fig. 2g). Cell division frequency at 14 days of culture on medium-A and medium-B was 23.7% and 24.5% respectively and colony formation frequency at 28 days of culture on medium-A and medium-B was10.9% and 11.2% respectively (Table 2).
Cell division and colony formation in liquid culture system
In general, protoplasts became elliptical and started to divide by the fifth day of culture. Cell division and colony formation were affected by medium composition. Medium-B was found to be more efficient for inducing cell divisions and colony formation than medium-A (Table 2).
In comparison with feeder-layer culture system, it was worth noting that all cell colonies derived from liquid culture system either in medium A or B were composed of large, highly vacuolated cells that appeared to be non-embryogenic (Fig. 2f), growth of those colonies was stopped at stage of 6 to 10-cells.
Re-initiation of cell suspensions from protoplast-derived cell colonies
Cell colonies derived from feeder-layer and liquid culture systems in medium-B were transferred into M2 liquid medium at a concentration of 3% (v/v). Following 1–2 months of culture, embryogenic and stable cell suspensions were established from PDCC derived from feeder-layer culture system. Morphological studies of these new suspension cultures showed no significant difference from the cv. Mas (AA) original ECS. PDCC derived from liquid culture in medium-B could remain alive in liquid M2 medium for more than 2 months, but the growth of those colonies was very slow and they showed non-embryogenic characteristics.
Effects of BAP, zeatin and TDZ on the formation of somatic embryo
After suspension in liquid M2 medium for 1 month, PDCC from both liquid and feeder-layer culture system in medium-B were transferred onto M3 medium with different concentrations of BAP, zeatin and TDZ for somatic embryogenesis. The M3 medium without cytokinin was used as control. PDCC derived from liquid culture in medium-B died after 10–15 days of culture, while PDCC derived from feeder-layer culture system in medium-B were increased in size, turned pale yellow after 7 days of culture and developed into mature bipolar embryo after 30 days of culture (Fig. 2h). Somatic embryos were formed within 1 month in all treatments with different frequencies. The frequency of embryo formation increased with BAP, zeatin and TDZ in the M3 medium. BAP at 4.4 μM, zeatin at 0.8 μM and TDZ at 0.4 μM were most effective compared to other concentrations tested (Table 3). While only 70 somatic embryos were obtained from 1 ml PCV PDCC in control medium, a maximum frequency of 7906 embryos/ml PCV PDCC was obtained with 0.4 μM TDZ, which was about 4-fold the frequency as with the best BAP concentration and 7.5-fold that with the best zeatin content and 150-fold the frequency as observed in control medium.
Plant regeneration
Mature bipolar embryos (Fig. 2h) derived from M3 medium with 0.4 μM TDZ were transferred onto M4 medium for germination, 44% of mature embryos were germinated after 30 days of culture (Fig. 2i). Poor root formation was observed from the base of plantlets regenerated in M4 medium. The regenerated plantlets with 2–3 cm shoots and poor root system were then transferred on RM for root further development. Addition of AC to the RM significantly promoted development of the root and shoot, and increased root number (9.5), root length (5.4 cm) and the height of plants (11.2 cm) compared to those cultured in control medium (5.7, 3.6 cm and 7.7 cm) (Table 4). The regenerated plants on MS medium with AC were more vigorous (Fig. 2j-1) than those on control MS medium (Fig. 2j-2) and they were easier to acclimatize.
Discussion
Effects of culture systems and media on cell division and colony formation from protoplasts
In this study we reported an efficient plant regeneration protocol from cell suspension-derived protoplasts of the dessert banana cv. Mas (AA) via somatic embryogenesis. We also showed that protoplasts of cv. Mas (AA) could divide and form cell colonies in liquid culture system (Table 2, Fig. 2g), contrasting previous reports that protoplasts of banana could not divide and form cell colonies when cultured in liquid medium (Panis et al. 1993; Assani et al. 2001). However, the cell colonies derived from liquid culture system did not develop further, while those derived from feeder-layer culture system could develop through a somatic embryogenesis pathway. It has been shown that the feeder-layer technique is always required for the protoplast culture of banana (Assani et al. 2002). One function of nurse cells, ECS of cv. Mas (AA), used in this study might be to secrete some soluble signal molecules that could stimulate the cell colonies derived from the protoplasts to become embryogenic cultures. ECS have been reported to secrete signal molecules such as endochitinase (De Jong et al. 1992; Egertsdotter and von Arnold 1998), and arabinogalactan proteins (AGPs) (Thompson and Knox 1998; Chapman et al. 2000), which were necessary for the development of somatic embryogenesis of Daucus carota, Picea abies and Cichorium hybrid ‘474’. It also has been showed that addition of carrot-seed AGPs to non-embryogenic cell suspensions of carrot resulted in the re-induction of embryogenic potential (Kreuger and van Holst 1993). The cell colonies derived from protoplasts of banana in liquid culture system were non-embryogenic and did not develop further, this problem might be resulted in no such signal molecules from nurse cells during liquid culture.
Our results showed that M2 liquid medium used for donor ECS material of cv. Mas (AA), with glucose as osmotic regulator, was suitable for protoplast culture. In this modified M2 medium, named medium-B, cell colonies were obtained from both liquid and feeder-layer culture systems. Similar results have been reported in various plant species, e.g. Vitis vinifera L. (Zhu et al. 1997) and Triticum aestivum (Xia et al. 1995). This could be explained by an easier adaptation of protoplasts to the original medium used for the donor tissues. In addition, in our studies, compared to medium-A, medium-B was cheaper and easier to manipulate.
Effects of BAP, zeatin and TDZ on somatic embryo formation of PDCC
It have indicated that embryo formation of many plant species required a high cytokinin supplemented medium, e.g. Coffea canephora (Hatanaka et al. 1991), Coronilla varia (Moyer and Gustine 1984), Medicago truncatula (Nolan et al. 1989), Thevetia peruviana (Kumar 1992) and Eleusina caracana (Eapen and George 1989). It also has been shown that cytokinin was essential for embryo formation of banana ECS (Nova et al. 1989; Dhedà et al. 1991; Ma 1991; Côte et al. 1996; Grapin et al. 1996). In protoplast culture of banana, BAP was also commonly used for somatic embryogenesis (Megia et al. 1993; Assani et al. 2001), but the conversion rate of microcalli or PDCC into somatic embryos was very low (about 2%). In this study, we found for the first time that TDZ at 0.4 μM was most effective and gave the highest frequency of embryo formation. Various reports have indicated that TDZ may act through a modulation of endogenous plant growth regulators (Visser et al. 1992; Hutchinson and Saxena 1996), either directly or as a result of induced stress (Murch and Saxena 1997). But the exact mechanism of TDZ to promote embryo formation of the PDCC needs to be investigated further.
Effects of AC on root development
Addition AC to the medium for root development is a common practice (Gould et al. 1991; Rasai et al. 1994; Ouma et al. 2004), but the exact mechanism for the promoting effects of AC on root development is still unknown. In this study, when the regenerated plantlets were cultured on MS medium with AC, the roots were more vigorous and more lateral roots were developed (Table 4, Fig. 2-j-1). Sanchez and San-Jose (1996) suggested that the stimulatory effects of AC on root development may involve: (1) the reduction of light intensity at the base of shoots, providing an environment conducive to the accumulation of auxin or cofactors, or both; and (2) the absorption of substances such as inhibitory phenolics and any excess auxin or cytokinin carried over from previous media. These effects may be true of our results.
The next stage of our investigation will be the extension of this protocol to produce somatic hybrids to generate novel genotypes between two Musa species.
Abbreviations
- AC:
-
Activated charcoal
- BAP:
-
6-benzylaminopurine
- 2,4-D:
-
2,4-dichlorophenoxyacetic acid
- ECS:
-
Embryogenic cell suspensions
- FDA:
-
Fluorescein diacetate
- Gln:
-
Glutamine
- IAA:
-
Indolacetic-3-acid
- MES:
-
2-N-morpholino ethanesulfonic acid
- NAA:
-
1-naphthaleneacetic acid
- PCV:
-
Packed cell volume
- PDCC:
-
Protoplast-derived cell colonies
- TDZ:
-
Thidiazuron
References
Assani A, Haïcour R, Wenzel G, Côte FX, Bakry F, Foroughi-Wehr B, Ducreux G, Aguillar ME, Grapin A (2001) Plant regeneration from protoplasts of dessert banana cv. Grande Naine (Musa spp., Cavendish sub-group AAA) via somatic embryogenesis. Plant Cell Rep 20:482–488
Assani A, Haïcour R, Wenzel G, Foroughi-Wehr B, Bakry F, Côte FX, Ducreux G, Ambroise A, Grapin A (2002) Influence of donor material and genotype on protoplast regeneration in banana and plantain cultivars (Musa spp.). Plant Sci 162:355–362
Assani A, Chabane D, Haïcour R, Bakry F, Wenzel G, Foroughi-Wehr B (2005) Protoplast fusion in banana (Musa spp.): comparison of chemical (PEG: polyethylene glycol) and electrical procedure. Plant Cell Tissue Org Cult 83:145–151
Assani A, Chabane D, Foroughi-Wehr B, Wenzel G (2006) An Improved protocol for microcallus production and whole plant regeneration from recalcitrant banana protoplasts (Musa spp.). Plant Cell Tissue Org Cult 85:257–264
Bakry F (1984) Choix du matériel à utiliser pour I’solement de protoplasts de bananier (Musa sp.). Friuts 39:449–452
Chapman A, Blervacq AS, Vasseur J, Hilbert JL (2000) Arabinogalactan protein in Cichorium somatic embryogenesis: effect of ß-glucosyl Yariv reagent and epitope localization during embryo development. Planta 211:305–314
Chu CC, Wang CC, Sun CS (1975) Establishment of an efficient medium for anther culture of rice through comparative experiments on the nitrogen source. Sci Sin 17:659–668
Côte FX, Domergue R, Monmarson S, Schwendiman J, Teisson C, Escalant JV (1996) Embryogenic cell suspensions from the male flower of Musa AAA cv Grande Naine Physiol Plant 97:285–290
Dhedà D, Dumortier F, Panis B, Vuylsteke D, De Langhe E (1991) Plant regeneration in cell suspension cultures of cooking banana cv. “Bluggoe” (Musa spp. ABB group). Fruits 46:125–135
De Jong AJ, Cordewener J, Lo Shiavo F, Terzi M, Vandekerckhove J, Van Kammen A, De Vries SC (1992) A carrot somatic embryo mutant is rescued by chitinase. Plant Cell 4:425–433
Eapen S, George L (1989) High frequency plant regeneration through somatic embryogenesis in finger millet (Eleusian carcana Gaertn.). Plant Sci 61:127–130
Egertsdotter U, von Arnold S (1998) Development of somatic embryos in Norway spruce. J Exp Bot 49:155–162
Escalant JV, Teisson C, Côte FX (1994) Amplified somatic embryogenesis from male flowers of triploid banana and plantain cultivars (Musa spp.). In Vitro Cell Dev Biol 30:181–186
Fock I, Collonnier C, Purwito A, Luisetti J, Souvannavong V, Vedel F, Servaes A, Ambroise A, Kodia H, Ducreux G, Sihachakr D (2000) Resistance to bacterial wilt in somatic hybrids between Solanum tuberosum and Solanum phureja. Plant Sci 160:165–176
Gould J, Banister S, Hasegawa O, Fahima M, Smith RH (1991) Regeneration of Gossypium hirsutum and G.barbadebse from shoot spex tissues for transformation. Plant Cell Rep 10:12–16
Grapin A, Schwendiman J, Teisson C (1996) Somatic embryogenesis in plantain banana. In Vitro Cell Dev Biol 32:66–71
Hatanaka T, Arakawa O, Yasuda T, Uchida N, Yamaguchi T (1991) Effect of plant growth regulators on somatic embryogenesis in leaf culture of Coffea canephora. Plant Cell Rep 10:179–182
Hutchinson MJ, Saxena PK (1996) Acetylsalicylic acid enhances and synchronizes thidiazuron-induced somatic embryogenesis in geranium (Pelargonium × Hortorum Bailey) tissue cultures. Plant Cell Rep 15:512–515
Kao KN, Michayluk MR (1975) Nutritional requirements for growth of Vicia hajastana cells and protoplasts at a very low population density in liquid media. Planta 126:105–110
Kreuger M, van Holst G (1993) Arabinogalactan proteins are essential in somatic embryogenesis of Daucus carota L. Planta 189:243–248
Kumar A (1992) Somatic embryogenesis and high frequency plantlet regeneration in callus cultures of Thevetia peruviana. Plant Cell Tissue Org Cult 31:47–50
Ma SS (1991) Somatic embryogenesis and plant regeneration from cell suspension culture of banana. In: Department of Agriculture, National Taiwan University (eds) Proceedings of symposium on tissue culture of horticultural crops. Taibei, Taiwan, 8, 9 March, 1988, pp 181–188
Matsumoto K, Oka S (1998) Plant regeneration from protoplasts of a Brazilian dessert banana (Musa spp., AAB group). Acta Hort 490:455–462
Matsumoto K, Vilarinhos AD, Oka S (2002) Somatic hybridization by electrofusion of banana protoplasts. Euphytica 125:317–324
Megia R, Haicour R, Rossignol L, Sihachakr D (1992) Callus formation from cultured protoplasts of banana (Musa sp.). Plant Sci. 85:91–98
Megia R, Hacour R, Tizroutine S, Bui Trang V, Rossignol L, Sihachakr D, Schwendiman J (1993) Plant regeneration from cultured protoplasts of the cooking banana cv. Bluggoe (Musa spp., ABB group). Plant Cell Rep 13:41–44
Morel G, Wetmore RH (1951) Fern callus tissue culture. Am J Bot 38:141–143
Moyer BG, Gustine DL (1984) Regeneration of Coronilla varia L. (crownvetch) plants from callus culture. Plant Cell Tissue Org Cult 3:143–148
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Murch SJ, Saxena PK (1997) Modulation of mineral and fatty acid profiles during thidiazuron mediated somatic embryogenesis in peanuts (Arachis hypogeae L.). J Plant Physiol 151:358–361
Nagata T, Takebe I (1970) Cell wall regeneration and cell division in isolated tobacco mesophyll protoplasts. Planta 92:301–308
Nolan KE, Rose RJ, Gost JR (1989) Regeneration of Medicago truncatula from tissue culture: increased somatic embryogenesis using explants from regenerated plant. Plant Cell Rep 8:279–281
Novak FJ, Afza R, Van Duren M, Perea-Dallos M, Conger BV, Tang XL (1989) Somatic embryogenesis and plant regeneration in suspension cultures of dessert (AA and AAA) and cooking (ABB) bananas (Musa spp.). Biotech 7:147–158
Ouma JP, Young MM, Reichert NA (2004) Rooting of in vitro regenerated cotton (Gossypium hirsutum L.) is influenced by genotype, medium composition, explant type and age. Afri J Biotech 3:313–318
Panis B, Wauwe AW, Swennen R (1993) Plant regeneration through direct somatic embryogenesis from protoplasts of banana (Musa spp.). Plant Cell Rep 12:403–407
Rasai S, Kantharajah AS, Dodd WA (1994) The effect of growth regulators, source of explants and irradiation on in vitro regeneration of Atemoya. Aust J Bot 42:333–340
Sanchez MC, San-Jose MC, Ballester A, Vieitez AM (1996) Requirements for in vitro rooting of Quercus robur and Qrubra rubra shoots derived from mature trees. Tree Physio 16:673–680
Thompson HJM, Knox JP (1998) Stage-specific responses of embryogenic carrot cell suspension cultures to arabinogalactan protein-binding ß-glucosyl Yariv reagent. Planta 205:32–38
Visser C, Qureshi JA, Gill R, Saxena P (1992) Morphoregulatory role of thidiazuron. Substitution of auxin and cytokinin requirement for the induction of somatic embryogenesis in geranium hypocotyls cultures. Plant Physiol 99:1704–1707
Wei YR, Huang XL, Li J, Huang X, Li Z, Li XJ (2005) Establishment of embryogenic cell suspension culture and plant regeneration of edible banana Musa acuminata cv. Mas (AA). Chin J of Biotech 21:58–65 (in Chinese)
Widholm JM (1972) The use of fluorescein diacetate and phenosafranine for determining viability of cultured plant cells. Stain Technol 47:189–194
Xia GM., Li ZY, Zhou AF, Guo GQ, Chen HM (1995) Plant regeneration of wheat protoplasts prepared from different composition of suspension culture. Chin J of Biotech 11:63–66 (in Chinese)
Yan GQ, Qian KX, Yan QS, Zhang XQ, Xue GP, Huang WG, Wu YF, Zhao YZ, Xue ZY, Huang J, Xu ZH, Wu P (2004) Use of asymmetric somatic hybridization for transfer of the bacterial blight resistance trait from Oryza meyeriana L. to O. sativa L. ssp. japonica. Plant Cell Rep 22:569–575
Zhu YM, Hoshino Y, Nakano M, Takahashi E, Mii M (1997) Highly efficient system of plant regeneration from protoplasts of grapevine (Vitis vinifera L.) through somatic embryogenesis by using embryogenic callus cultures and activated charcoal. Plant Sci 123:151–157
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (30400287), Guangdong Natural Science Foundation of China (04300538, 06023159, and 06300470), Guangdong Scientific and Technical Foundation (2006B20101014) and Guangzhou Scientific and Technical Foundation (2006Z3-E0281).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Xiao, W., Huang, XL., Huang, X. et al. Plant regeneration from protoplasts of Musa acuminata cv. Mas (AA) via somatic embryogenesis. Plant Cell Tiss Organ Cult 90, 191–200 (2007). https://doi.org/10.1007/s11240-007-9241-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-007-9241-4