
Abstract
Abnormal platelet reactivity is associated with recurrent ischemia and bleeding following percutaneous coronary intervention (PCI). Protease-activated receptor-1 (PAR1), encoded by F2R, is a high affinity thrombin receptor on platelets and the target of the antiplatelet drug vorapaxar. The intronic single nucleotide polymorphism F2R IVS-14 A/T affects PAR1 receptor density and function. We hypothesized that carriers of the T allele, who have been shown to have decreased platelet reactivity, would be at lower risk for thrombotic events, but higher risk for bleeding following PCI. Using BioVU, the Vanderbilt DNA repository linked to the electronic medical record, we studied 660 patients who underwent PCI for unstable or stable coronary artery disease. Primary outcome measures were major adverse cardiovascular events (MACE, composite of revascularization, MI, stroke, death) and bleeding (assessed by Bleeding Academic Research Consortium scale) over 24 months. The minor allele (T) frequency was 14.8 %. There were no genotypic differences in the frequency of MACE (33.7, 28.8, and 31.6 % for A/A, A/T, and T/T respectively, P = 0.50) or bleeding (15.7, 14.7, and 18.8 % for A/A, A/T, and T/T respectively, P = 0.90). In a Cox regression model, fully adjusted for age, race, sex, BMI, and smoking status, carrying a T allele was not associated with MACE (HR 1.19, 95 % CI 0.89–1.59, P = 0.23) or bleeding (HR 0.73, 95 % CI 0.37–1.4, P = 0.34). In conclusion, in our population, F2R IVS-14 PAR1 variability does not affect risk of MACE or bleeding following PCI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Dual antiplatelet therapy with aspirin and a P2Y12 inhibitor is recommended for patients following a myocardial infarction (MI) or percutaneous coronary intervention (PCI). On-treatment platelet reactivity, when abnormally high or low, is associated with recurrent ischemia and bleeding [1]. The bulk of research has focused on variability in platelet reactivity during treatment with the platelet P2Y12 inhibitor clopidogrel. Specifically, genetic variability in the CYP2C19 gene, which metabolizes the clopidogrel pro-drug, has been associated with clinical outcomes in patients on dual antiplatelet therapy [2–5]. However, inter-individual variability in platelet activation pathways upstream, downstream, or apart from P2Y12 likely also contributes to overall platelet reactivity and may impact the risk of ischemic or bleeding events [6].
Thrombin, the major protease in the coagulation cascade, is also the most potent platelet activator and has a crucial role early in the platelet aggregation cascade. Thrombin works by activation of the G protein-coupled protease-activated receptors 1 and 4 (PAR1 and PAR4) on human platelets to initiate signaling cascades. Subsequent increases in [Ca]i secretion and activation of integrins initiate platelet aggregation [7, 8]. Activation of human platelets by thrombin is mediated by activation of both PAR1 and PAR4, with PAR1 being the high affinity thrombin receptor [7]. Recently, the drug vorapaxar, a PAR1 antagonist, was approved as adjunctive antiplatelet therapy.
Inter-individual variability in the platelet response to PAR1 activation has been recognized, using the PAR1-agonist peptide SFLLRN. Each individual’s responsiveness to SFLLRN is stable over time, suggesting a genetic factor [9]. In a study of 100 healthy male volunteers, Dupont and others examined a single SNP in the PAR1 gene that is 14 nucleotides upstream of the exon 2 start site (IVS-14 A/T, rs168753) [10]. The authors found that the T allele is associated with decreased expression of PAR1 on the platelet surface and a decreased functional response to PAR1 activation. A further study investigated rs168753 in patients with coronary artery disease treated with clopidogrel. The authors of this study found higher platelet reactivity associated with homozygotes for the A allele despite clopidogrel therapy [11]. Studies looking at platelet reactivity based on P2Y12 profile have demonstrated that patients with higher platelet reactivity have increased rates of bleeding, and those who have lower reactivity have decreased bleeding [12–15]. These data suggest that individuals homozygous for the A allele at the rs168753 loci of F2R might be at increased risk of subsequent ischemic cardiovascular events. Accordingly individuals carrying the T allele would be postulated to have increased risk of bleeding. However, an association of this F2R polymorphism with clinical events is unknown. We hypothesized that rs168753 genotype is associated with risk of major adverse cardiovascular events and bleeding during dual antiplatelet therapy following MI or PCI.
Methods
Subject identification
The Vanderbilt University Medical Center electronic health record (EHR) includes comprehensive information on ~1.7 million patients; BioVU, the Vanderbilt DNA biobank, includes de-identified DNA samples from more than 120,000 individuals. These samples are linked to a de-identified form of the EHR known as the Synthetic derivative (SD). From the BioVU database we identified individuals who were started on clopidogrel therapy after a MI or PCI. International Classification of Disease, 9th edition (ICD-9), Current Procedural Terminology (CPT) codes, laboratory values, medication orders, and natural-language processing in physician notes were used to screen the SD and identify these patients as described previously [16]. Major adverse cardiovascular events (MACE) were defined as the occurrence of MI, stroke, revascularization, or death in the 24 months following the initial event. The occurrence of a secondary MACE endpoint of stent thrombosis was also obtained during review.
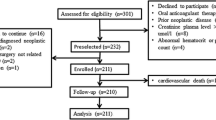
The occurrence of bleeding within 24 months after the index event was screened using previously validated ICD-9 codes indicative of bleeding [17] and natural language processing in physician notes. Manual chart review of the SD was performed for each case to adjudicate bleeding and stratify severity (Fig. 1). The manual review was performed separately by two study investigators (L.T. and E.F.) who were blinded to genotype, and any discrepancies were adjudicated by a third reviewer (J.C.). Bleeding severity was stratified using the Bleeding Academic Consortium (BARC) scale [18]. The BARC scale is defined as follows: Type 0 (no bleeding), BARC 1 (bleeding that is not actionable), Type 2 (overt bleeding that does not meet criteria for Types 3 and above), Type 3a (bleeding with hemoglobin decrease of 3–5 g/dL or need of transfusion), Type 3b (bleeding with hemoglobin decrease >5 g/dL, tamponade, need for surgical intervention, or need for vasoactive substances), Type 3C (intracranial hemorrhage or intraocular compromising vision), Type 4 (Coronary Artery Bypass related bleeding), or Type 5 (fatal bleeding).

Algorithm for Screening Cases for the presence of bleeding. Potential bleeding cases were identified by ICD-9 code and natural language processing, then were either confirmed or excluded by manual review. To meet inclusion criteria, the bleeding episode had to occur during treatment with clopidogrel within 24 months of the index MI or PCI
The patient cohort in this study was based on a previous case–control study evaluating an association of MACE with genetic factors [16]. The occurrence of bleeding was performed as an analysis within this cohort. In between performing the analysis for MACE and bleeding, 81 patients opted to have their DNA removed from BioVU; therefore, the bleeding analysis was performed with a smaller cohort of 587 patients.
Sample analysis
DNA was extracted from stored samples using a Gentra Systems Autopure LV automated DNA extraction system Genotyping was conducted using Applied Biosystems (ABI, Foster City, CA, USA) TaqMan Assays-on-Demand. PCR for the rs1126709 and rs168753 PAR1 SNPs was carried out in 384-well plates in 5 ml reactions containing 0.125 ml 20X Assays-on-Demand probe/primer mix, 5 ng of genomic DNA and TaqMan Universal PCR Mastermix according to the manufacturer’s recommendations. Products were scanned on the ABI 7900HT instrument.
Statistical analysis
The cumulative incidence of the primary outcomes MACE and Bleeding by SNP (rs168753) genotype was estimated with the Kaplan–Meier product limit estimator, and unadjusted comparisons were performed with log rank test. Cox proportional hazards model assuming an additive model was performed and adjusted for the following covariates: age, gender, race (European American, African American, other/unknown), body mass index (BMI), and smoking status (current, ever, never). The secondary endpoint stent thrombosis was included in the analysis. One-way analysis of variance was used to assess differences in covariates between genotypes.
Results
Using a phenotypic algorithm as previously described, 660 patients were identified who were started on clopidogrel therapy after MI and/or PCI. Minor allele frequency of the T allele was 16.1 % for a Caucasian population and 8.3 % in an African American population based on HapMap data, while we found the MAF within our population of subjects undergoing PCI to be 14.8 %.
Demographic, clinical characteristics, and the frequency of MACE and bleeding are depicted in Table 1. Carriers of the T allele had slightly lower rates of diabetes. Individuals homozygous for the T allele had lower rates of hypertension. In our patient cohort, 214 subjects had a major adverse cardiovascular event. Stent thrombosis occurred in 11 of these subjects. Of the 587 subjects available for the bleeding analysis, the outcome of bleeding occurred in 90 subjects (15.3 %). Procedure-related bleeding in our cohort (4.8 %) was similar to large published observational studies (3.1 %) [19]. While our overall rate of bleeding at 2 years was higher than the reported rate of 9.9 % at one year [20], given the extra year of exposure to clopidogrel, our bleeding rate is comparable. Our rate of BARC > 2 bleeding is similar to the reported 5.4 % [20]. There were no statistically significant differences in the frequency of MACE, stent thrombosis, or bleeding between genotypes. Bleeding was not a significant contributor to MACE, as severe bleeding (BARC > 2) was not significantly different between patients who had MACE or did not have MACE (4.27 vs. 4.21 %, respectively, P = 0.31).
A Kaplan–Meier curve of the primary end-point of MACE for PAR1 rs168753 demonstrated that carriers of a T allele had similar event rates compared to subjects without this allele (P = 0.50) (Fig. 2). Analysis of bleeding similarly was not different between genotypic groups (P = 0.90) (Fig. 2). In a Cox regression model, fully adjusted for age, race, sex, BMI, and smoking status, carrying a T allele was not associated with MACE (HR 1.19, 95 % CI 0.89–1.59, P = 0.23) or bleeding (HR 0.73, 95 % CI 0.37–1.4, P = 0.34).

MACE-Free survival by genotype
Discussion
Our study has demonstrated that the F2R IVS-14 polymorphism in the gene encoding PAR1, which is known to affect platelet reactivity, does not contribute to risk of major adverse cardiovascular events or bleeding in patients treated with clopidogrel following MI or PCI (Fig. 3).

Bleed-free survival by GENOTYPE
It is well established that high on treatment platelet reactivity is associated with recurrent adverse cardiovascular events after PCI. In 2006 a candidate gene approach performed in 28 healthy humans subjects given clopidogrel first reported that a loss of function polymorphism involved in the metabolism of clopidogrel (2CYPC19*2) was associated with high on-treatment platelet reactivity [21]. Subsequent studies in patients undergoing PCI demonstrated that CYPC19*2 loss of function polymorphisms associate with recurrent adverse cardiovascular events [16, 22–24]. However, based on modeling in healthy subjects, CYP2C19 genotype only accounts for 4–11 % of the variability of response to clopidogrel [23]. More recently a study that measured the active metabolite of clopidogrel and excluded subjects with polymorphisms in CYP2C19, CYP3A5, ABC1, and PON1, while rigorously controlling for other demographics that influence platelet reactivity, concluded that unidentified factors contribute to high on-treatment platelet reactivity [25]. Accordingly, the search for additional polymorphisms that contribute to high on-treatment platelet reactivity has been underway. The GIFT-EXOME study used whole genome sequencing in 392 subjects treated with clopidogrel from the GRAVITAS study and found that three separate genomic loci were associated with on-treatment platelet reactivity [26].
In patients being treated with antiplatelet agents, the benefit at reducing ischemia is counterbalanced by an increased risk of bleeding. Newer and more potent antiplatelet agents carry even higher risks of bleeding. Some have suggested that there is a “ceiling effect” in antiplatelet agents, whereby there is an upper limit in ability to reduce ischemic events, and attempts to reduce ischemia further only lead to higher rates of bleeding [1, 27, 28]. As current antiplatelet medications, particularly the more potent P2Y12 antagonists, are quite effective at reducing MACE, it may be prudent to place more attention on safety measures. Bleeding events contribute to both morbidity and mortality in patients on long term antiplatelet therapy [29]. This may be due to direct complications from bleeding or its treatment or the need for premature antiplatelet drug discontinuation. Whereas numerous studies have explored the association of high platelet reactivity with ischemic events, there has been considerable less attention to the effects of low platelet reactivity on bleeding [1]. Small observational studies in patients following PCI have suggested a relationship between low on-treatment platelet reactivity and bleeding [12, 13, 15]. Large observational studies, including GRAVITAS and ARCTIC did not demonstrate such an association [5, 30]. However, differences in bleeding rates and definitions between studies limit the ability to make determinations.
Thrombin is the most potent platelet activating factor and works primarily through the protease-activated receptor 1 (PAR1), which is encoded by F2R. The effects of PAR1 in the platelet activation cascade precede those of the platelet P2Y12 receptor, such that inhibition of PAR1 has further anti-aggregatory effects to inhibition of P2Y12 [31]. Furthermore, P2Y12 activation acts in a positive feedback loop on the PAR1 receptor [31]. Accordingly, P2Y12 antagonists inhibit both thrombin and PAR1 agonist peptide stimulation of human platelets [32, 33]. Thus, it would be anticipated that combining P2Y12 and PAR1 antagonists would have synergistic antiplatelet effects that could have significant clinical implications. The addition of vorapaxar, a recently approved PAR1 antagonist, to dual antiplatelet therapy consisting of aspirin and clopidogrel in patients undergoing PCI for acute coronary syndromes did not improve ischemic outcomes and actually caused excess intracranial bleeding [34]. However, in a secondary prevention trial the addition of vorapaxar to aspirin and clopidogrel did reduce ischemic events in patients at high risk of atherothrombotic events, but did so at the cost of increasing intracranial hemorrhage [35]. Subgroup analyses of this trial involving the highest risk patients, those with diabetes and prior MI, showed that these populations had even greater reduction in ischemic events or death with vorapaxar [36, 37]. Furthermore, Vorapaxar added to standard dual antiplatelet therapy also reduced the occurrence of early and late stent thrombosis [38].
The IVS-14 A/T single nucleotide polymorphism in F2R (rs168753) modulates PAR1 expression and activity such that carriers of the T allele have decreased platelet activation in response to thrombin receptor agonists. While the polymorphism occurs in an intronic, non-coding segment, it is thought that it may have an effect on mRNA processing rate and transcription efficiency, leading to altered protein expression [10]. Dupont et al. found that biphasic aggregation occurred with a lower dose of PAR1 agonist SFFLRN in A/A than A/T and T/T subjects combined (9.1 ± 2.1 vs. 10.9 ± 3.0 μM, respectively, P = 0.003) [10]. The authors further reported that one of the homozygous T/T subjects required a very high SFFLRN concentration of 19 μM to achieve biphasic aggregation, suggesting that carrying two mutant alleles may lead to further reduction in platelet reactivity. Smith et al. showed that the F2R polymorphism conferred a difference in platelet aggregation to the thrombin receptor agonist protein even in the setting of clopidogrel exposure [11]. Based on these preclinical data along with the reported clinical effects of vorapaxar, we hypothesized that carriers of the T allele, whose reduced platelet reactivity is akin to partial PAR1 inhibition, might have decreased rates of ischemia, but increased rates of bleeding following MI or PCI. Moreover, as the effects of PAR1 are upstream to P2Y12, we anticipated PAR1 variability to have clinical implications even in the setting of P2Y12 blockade with clopidogrel. In our population, however, we did not find an association of the F2R polymorphism with risk of MACE or bleeding. Our results suggest that the overall effects of the polymorphism on platelet reactivity are not great enough to translate to clinically significant outcomes.
A strength of our current study is the well characterized patient cohort and the technique of using a de-identified medical record linked to a genetic databank. Such an approach has significant promise as a platform for large-scale studies correlating genetic variability with clinical outcomes. One limitation of the current study is the relatively low sample size, limiting our power to detect small differences in clinical endpoints. However, the argument could be made that significant genotype-phenotype differences that have the potential to change practice have already been found in this cohort. For example, the CYP2C19*2 loss of function polymorphism was associated with recurrent cardiovascular events [16]. A number of patients opted out of the DNA databank in between performing the ischemia and bleeding analyses, and their data thus had to be excluded. Within this smaller cohort, bleeding (defined as BARC any) only occurred in 90 patients (15.3 %), further limiting the power to detect small differences in bleeding frequency between genotypes. An alternative approach with greater statistical power would be to design a case–control study based on the presence or absence of bleeding and then to test for differences in genotype frequency. A recently published study on the pharmacogenetics of warfarin-related bleeding used such a method [39]. Finally, a limitation of the study is the inability to perform platelet function or biochemical assays in the subjects, which would provide further insight into the biologic effects of F2R variability. For example, clinical bleeding in individuals carrying the T allele could perhaps be mitigated by compensatory up-regulation of other platelet-activating pathways.
As the clinical use of vorapaxar increases, further research into the effects of PAR1 genetic variability is essential. Such investigation will help guide therapy to ensure optimal reduction of ischemic events with reduced risk of bleeding. In this era of widespread interest in personalized medicine, a genome-guided approach to rational drug therapy is proposed to eventually become standard of care. It is also clear that CYP2C19 variability, while an important determinant of platelet response to clopidogrel, is but one of a number of genes that contribute to platelet reactivity.
In conclusion, the IVS-14 F2R genotype, which affects platelet reactivity in vitro, did not affect risk of major adverse cardiovascular events or bleeding in our study population. Future large-scale genetic studies, such as the International Clopidogrel Platelet Consortium, will be important to discover novel genetic factors that associate with risks and benefits of cardiovascular therapy.
References
Tantry US, Bonello L, Aradi D et al (2013) Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol 62:2261–2273
Trenk D, Hochholzer W, Fromm MF et al (2008) Cytochrome P450 2C19 681G > A polymorphism and high on-clopidogrel platelet reactivity associated with adverse 1-year clinical outcome of elective percutaneous coronary intervention with drug-eluting or bare-metal stents. J Am Coll Cardiol 51:1925–1934
Harmsze A, van Werkum JW, Bouman HJ et al (2010) Besides CYP2C19*2, the variant allele CYP2C9*3 is associated with higher on-clopidogrel platelet reactivity in patients on dual antiplatelet therapy undergoing elective coronary stent implantation. Pharmacogenet Genomics 20:18–25
Simon T, Verstuyft C, Mary-Krause M et al (2009) Genetic determinants of response to clopidogrel and cardiovascular events. N Engl J Med 360:363–375
Collet JP, Cuisset T, Range G et al (2012) Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N Engl J Med 367:2100–2109
Bray PF (2007) Platelet hyperreactivity: predictive and intrinsic properties. Hematol Oncol Clin North Am 21:633–645, v–vi
Coughlin SR (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 3:1800–1814
Leger AJ, Covic L, Kuliopulos A (2006) Protease-activated receptors in cardiovascular diseases. Circulation 114:1070–1077
Lasne D, Krenn M, Pingault V et al (1997) Interdonor variability of platelet response to thrombin receptor activation: influence of PlA2 polymorphism. Br J Haematol 99:801–807
Dupont A, Fontana P, Bachelot-Loza C et al (2003) An intronic polymorphism in the PAR-1 gene is associated with platelet receptor density and the response to SFLLRN. Blood 101:1833–1840
Smith SM, Judge HM, Peters G et al (2005) PAR-1 genotype influences platelet aggregation and procoagulant responses in patients with coronary artery disease prior to and during clopidogrel therapy. Platelets 16:340–345
Sibbing D, Schulz S, Braun S et al (2010) Antiplatelet effects of clopidogrel and bleeding in patients undergoing coronary stent placement. J Thromb Haemost 8:250–256
Cuisset T, Cayla G, Frere C et al (2009) Predictive value of post-treatment platelet reactivity for occurrence of post-discharge bleeding after non-ST elevation acute coronary syndrome. Shifting from antiplatelet resistance to bleeding risk assessment? EuroIntervention 5:325–329
Campo G, Parrinello G, Ferraresi P et al (2011) Prospective evaluation of on-clopidogrel platelet reactivity over time in patients treated with percutaneous coronary intervention relationship with gene polymorphisms and clinical outcome. J Am Coll Cardiol 57:2474–2483
Parodi G, Bellandi B, Venditti F et al (2012) Residual platelet reactivity, bleedings, and adherence to treatment in patients having coronary stent implantation treated with prasugrel. Am J Cardiol 109:214–218
Delaney JT, Ramirez AH, Bowton E et al (2012) Predicting clopidogrel response using DNA samples linked to an electronic health record. Clin Pharmacol Ther 91:257–263
Cunningham A, Stein CM, Chung CP, Daugherty JR, Smalley WE, Ray WA (2011) An automated database case definition for serious bleeding related to oral anticoagulant use. Pharmacoepidemiol Drug Saf 20:560–566
Mehran R, Rao SV, Bhatt DL et al (2011) Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation 123:2736–2747
Rao SV, Dai D, Subherwal S et al (2012) Association between periprocedural bleeding and long-term outcomes following percutaneous coronary intervention in older patients. JACC Cardiovasc Interv 5:958–965
Ndrepepa G, Schuster T, Hadamitzky M et al (2012) Validation of the Bleeding Academic Research Consortium definition of bleeding in patients with coronary artery disease undergoing percutaneous coronary intervention. Circulation 125:1424–1431
Hulot JS, Bura A, Villard E et al (2006) Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 108:2244–2247
Mega JL, Close SL, Wiviott SD et al (2009) Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med 360:354–362
Shuldiner AR, O’Connell JR, Bliden KP et al (2009) Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA 302:849–857
Stone GW, Witzenbichler B, Weisz G et al (2013) Platelet reactivity and clinical outcomes after coronary artery implantation of drug-eluting stents (ADAPT-DES): a prospective multicentre registry study. Lancet 382:614–623
Frelinger AL 3rd, Bhatt DL, Lee RD et al (2013) Clopidogrel pharmacokinetics and pharmacodynamics vary widely despite exclusion or control of polymorphisms (CYP2C19, ABCB1, PON1), noncompliance, diet, smoking, co-medications (including proton pump inhibitors), and pre-existent variability in platelet function. J Am Coll Cardiol 61:872–879
Price MJ, Carfson AR, Murray SS et al (2012) First pharmacogenomic analysis using whole exome sequencing to identify novel genetic determinants of clopidogrel response variability: results of the Genotype Information and Functional Testing (GIFT) Exome Study. J Am Coll Cardiol 59:E9
Bhatt DL (2009) Prasugrel in clinical practice. N Engl J Med 361:940–942
Menown IB (2011) Aspirin, P2Y12 blockers, cilostazol, PAR-1 blockers and emerging antiplatelet therapies: can biomarkers guide clinical development and practice? Biomark Med 5:1–3
Chhatriwalla AK, Amin AP, Kennedy KF et al (2013) Association between bleeding events and in-hospital mortality after percutaneous coronary intervention. JAMA 309:1022–1029
Price MJ, Berger PB, Teirstein PS et al (2011) Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA 305:1097–1105
Storey RF, Kotha J, Smyth SS et al (2014) Effects of vorapaxar on platelet reactivity and biomarker expression in non-ST-elevation acute coronary syndromes. The TRACER Pharmacodynamic Substudy. Thromb Haemost 111:883–891
Cleator JH, Duvernay MT, Holinstat M et al (2014) Racial differences in resistance to P2Y12 receptor antagonists in type 2 diabetic subjects. J Pharmacol Exp Ther 351:33–43
Behan MW, Fox SC, Heptinstall S, Storey RF (2005) Inhibitory effects of P2Y12 receptor antagonists on TRAP-induced platelet aggregation, procoagulant activity, microparticle formation and intracellular calcium responses in patients with acute coronary syndromes. Platelets 16:73–80
Tricoci P, Huang Z, Held C et al (2012) Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med 366:20–33
Morrow DA, Braunwald E, Bonaca MP et al (2012) Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med 366:1404–1413
Scirica BM, Bonaca MP, Braunwald E et al (2012) Vorapaxar for secondary prevention of thrombotic events for patients with previous myocardial infarction: a prespecified subgroup analysis of the TRA 2 degrees P-TIMI 50 trial. Lancet 380:1317–1324
Cavender MA, Scirica BM, Bonaca MP et al (2015) Vorapaxar in patients with diabetes mellitus and previous myocardial infarction: findings from the thrombin receptor antagonist in secondary prevention of atherothrombotic ischemic events-TIMI 50 trial. Circulation 131:1047–1053
Bonaca MP, Scirica BM, Braunwald E et al (2014) Coronary stent thrombosis with vorapaxar versus placebo: results from the TRA 2 degrees P-TIMI 50 trial. J Am Coll Cardiol 64:2309–2317
Kawai VK, Cunningham A, Vear SI et al (2014) Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics 15:1973–1983
Author information
Authors and Affiliations
Corresponding author
Additional information
Work supported by Vanderbilt Institute for Clinical and Translational Research Award to JHC.
Rights and permissions
About this article

Cite this article
Friedman, E.A., Texeira, L., Delaney, J. et al. Evaluation of the F2R IVS-14A/T PAR1 polymorphism with subsequent cardiovascular events and bleeding in patients who have undergone percutaneous coronary intervention. J Thromb Thrombolysis 41, 656–662 (2016). https://doi.org/10.1007/s11239-015-1285-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-015-1285-4