
Abstract
The objective of this study was to use genome-wide association approach and pooled DNA strategy to search for new genomic loci associated with inter-individual differences in platelet reactivity in the diabetic patients during acetylsalicylic acid (ASA) treatment. Study cohort consisted of 297 diabetic patients who had been taking ASA (75 mg daily) for at least 3 months. We tested association of single nucleotide polymorphisms (SNPs) genotyped using high density microarray platform with several platelet reactivity assays, followed by individual genotyping of most significant SNPs identified in the microarray genomic scan. The highest statistical significance (p value of 0.0001–0.008 in individual genotyping) was observed for SNP located within the regulatory G-protein signaling (RGS) 7 gene (rs2502448) using recessive genetic model. The diabetic patients on ASA treatment and homozygotes for its minor allele were characterized by increased odds ratio of at 3.45 (confidence interval: 1.82–6.53) for high on ASA platelet reactivity (i.e. impaired ASA response) when compared with homozygotes for wild-type allele. The genome-wide approach might provide an opportunity to identify novel candidate genes and pathways related to platelet activation in diabetic patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acetylsalicylic acid (ASA) remains the primary antiplatelet drug used in patients at high risk for cardiovascular disease, in particular in type 2 diabetes mellitus (T2DM). Despite advances in the antiplatelet therapy, patients with T2DM continue to experience a higher risk of ischemic events than non-diabetic patients. Moreover, it was reported that T2DM patients display high platelet reactivity, elevated levels of platelet thromboxane, and that ASA therapy was associated with higher platelet reactivity in this group of patients [1]. ASA is an effective inhibitor of platelet thromboxane A2 (TXA2) synthesis by cyclooxygenase type 1 (COX1), nevertheless it is often considered a relatively weak platelet inhibitor because of its limited effects on aggregation in the presence of high concentrations of other agonists such as adenosine diphosphate (ADP) or collagen. This may account for some of the inter-individual variability in response to these stimulants, which activate platelets through both TXA2-dependent and -independent pathways [2]. Several single nucleotide polymorphisms (SNPs) could be responsible for at least some apparent inter-individual differences in platelet reactivity in diabetic population treated with ASA. The results from our own recent study using targeted multiplexing of previously recognized genomic loci associated with altered platelet reactivity suggest that 4 genomic loci (TXBA2R, ADRA2A, PLA2G7, 9p21.3) may contribute to platelet reactivity (as measured by PFA-100 assay) in this population [3]. As an economically feasible alternative of individual genome wide association study (GWAS) approach for discovery of new genomic markers of variable ASA response in T2DM, we used the previously established DNA pooling strategy. Pooling allows for equal amounts of DNA to be combined to form sample pools from patients with different platelet reactivity during ASA treatment, which are then genotyped to estimate allele frequency differences for each SNP. This information is then used for confirmatory association studies for a limited number of identified candidate SNPs. Thus, the main goal of this study was to use GWAS and pooled DNA samples strategy to search for new loci associated with the altered platelet reactivity [selected on the basis of the inter-individual differences in the collagen/epinephrine (CEPI)-closure time (CT) measurement of the platelets reactivity] in the population of exclusively T2DM patients.
Methods
Patient population and study design
The study was approved by the ethics committee of the Medical University of Warsaw and conducted in accordance with the current version of the Declaration of Helsinki. The genotyping part of the study was reviewed and approved by the Institutional Review Board of Penn State Hershey Medical Center (Hershey, PA, USA). The study subjects were recruited consecutively from patients with T2DM (participating in a multi-center, prospective, randomized, and open-label AVOCADO [Aspirin vs/or Clopidogrel in Aspirin-resistant diabetics inflammation outcomes] study presenting to the outpatient clinic of the Central Teaching Hospital of the Medical University of Warsaw. The full characterization of the study population, including the inclusion and exclusion criteria were published previously [3]. Briefly, the Caucasian subjects with T2DM were recruited who, at the time of enrollment, had been taking ASA tablets at the dose of 75 mg per day for at least 3 months for primary or secondary prevention of myocardial infarction (MI). No clopidogrel or antiplatelet drugs other than ASA were used in any of the investigated patients. All patients had been taking oral antidiabetic agents and/or insulin for at least 6 months; diet-controlled diabetic patients were not included. Compliance to ASA therapy at the study entry was determined based upon the patient’s own statement and serum thromboxane B2 (S-TXB2) level measurement.
Blood sample and assay procedures
Blood samples were taken in the morning 2–3 h after the last ASA dose. Regular laboratory testing was performed using standard laboratory techniques. The concentration of functional epitope of the von Willebrand factor (vWF) molecule (vWF: Ag) was measured in citrate plasma samples using an enzyme immunoassay kit according to the manufacturer’s instructions (vWF Activity Kit, American Diagnostica Inc., USA). S-TXB2 was measured also with an enzyme immunoassay kit according to the manufacturer’s instructions (Cayman Chemicals, Ann Arbor, MI, USA). The compliance with ASA treatment was defined by the S-TXB2 levels below 7.2 ng/mL [1].
Platelet function analysis
VerifyNow-Aspirin Assay (Accumetrics, San Diego, CA, USA) is a turbidimetric-based optical detection system, which measures platelet-induced agglutination as the increase in light transmittance in response to arachidonic acid, and converts luminosity transmittance units into aspirin reaction units (ARU) for VerifyNow. According to the manufacturer, ARU ≥ 550 indicates no effect of ASA on platelet aggregation, whereas ARU < 550 indicates platelet dysfunction due to inhibition of the COX1-dependent pathway [4].
PFA-100-assay (Dade-Behring International, Inc., Newark, DE, USA) is based on the aspiration of the citrated whole blood at a high shear rate through a small aperture cut in a membrane, coated with 2 μg of type I collagen and either 10 μg epinephrine (EPI) bitartrate or 50 μg adenosine diphosphate (ADP). The instrument records the time necessary for occlusion of the aperture, defined as CT, which is indicative of platelet reactivity in the whole blood sample. Based on our own and other previous reports, we applied three different cut-off values for high platelet reactivity in the CEPI-CT assay, but we did not specify such a point for collagen/adenosine diphosphate (CADP)-CT. In the first approach, adequate platelet inhibition with ASA was defined as CEPI-CT ≥ 165 s, and in the second as CEPI-CT ≥ 193 s (the manufacturer’s suggested lower limit of the normal range for aspirin-free healthy controls) [5]. The maximum CT given for PFA-100 is 300 s and is equivalent to non-occlusion [6]. Thus, patients with CEPI-CT values ≥ 300 s were defined as an alternative population with adequate platelet inhibition [7].
DNA extraction and quantification
DNA was extracted from whole blood samples by using the membrane ultrafiltration method with Fuji MiniGene 80 extractor (FujiFilm Life Sciences distributed by Autogene, Holliston, MA, USA). Two DNA pools were constructed (high on ASA platelet activity patients defined by PFA-100 CEPI-CT < 193 s, and patients with not high on ASA platelet activity defined as PFA-100 CEPI-CT ≥ 193 s) by combining equimolar amounts of DNA from each individual. The final pools were concentrated to at least 200 ng using Microcon YM-100 Centrifugal Filter Units (Millipore Corporation, MA). The final DNA concentrations in case and control pools was confirmed with the PicoGreen method (PicoGreen dsDNA Quantitation Reagent Kit, Molecular Probes Inc., Eugene, OR, USA).
Microarray genotyping
Genome-wide genotyping was performed using Illumina HumanOmni2.5-Quad microarrays (Illumina Inc., San Diego, CA, USA) using Infinium LCG® protocol and according to the manufacturer’s recommendations. Microarrays were scanned using Illumina HiScan system by Beckman Coulter Genomics (Morrisville, NC, USA) and raw data were extracted for statistical analysis with Illumina GenomeStudio v.2010.3 software (Illumina Inc., San Diego, CA, USA). Approximation of allele frequencies for each marker was produced on the basis of raw data by the Illumina GenomeStudio software and based on the internal calibration and normalization procedures [8]. Generation of minor and major allele frequencies was performed using the self-normalization algorithm of the genotyping module of the GenomeStudio software.
Individual SNP genotyping
Genotyping for selected markers (see Supplemental Table 1) in individual DNA samples was performed at Children’s Hospital Boston using a custom Sequenom iPLEX assay in conjunction with the Mass ARRAY platform (Sequenom Inc., La Jolla, CA, USA). One panel of SNP markers was designed using Sequenom Assay Design 3.2 software (Sequenom Inc., La Jolla, CA, USA).
Analyses of the pooling experiment
The analysis of the pooled frequency data was performed in 3 stages: (1) filtering of the raw allele frequency data for all SNPs on the microarray, (2) SNP ranking by statistical significance, and (3) final ranking by a versatile gene-based test for GWAS (VEGAS). The test statistic for the remaining allele differences was based on p-values estimated using the modified Chi2 statistics (Tcomb test) which combines experimental and sampling errors, a general description of which has been extensively validated previously [9]. VEGAS approach incorporates information from a full set of markers within a gene boundaries and accounts for linkage disequilibrium (LD) between markers by using simulations from the multivariate normal distribution [10]. This test, which only requires the uploading of markers with their association p-values, was performed remotely by the VEGAS software available through Internet in Genetics and Population Health Division, Queensland Institute of Medical Research, Brisbane, Australia (http://genepi.qimr.edu.au/general/softwaretools.cgi).
Statistical analysis of the individual genotyping results
Power analysis
The calculations of the approximate power statistics and sample size for pooling studies were performed using CaTS—power calculator for two stage association studies [11]. Based on the population incidence of high on ASA platelet activity in diabetic population (30 %), we calculated that the power to detect an association at p < 10−7 (genetic risk of at least 2.0 for an allele with frequency 50 %, with the minimum of 240 enrolled subjects). Results from Power and Sample Size Calculator v.03.14 provided a secondary power assessment, as described previously [12]. For the power analysis in the individual genotyping, we calculated that for the expected change in the minor allele frequency of variant allele in subjects with high on ASA platelet activity by 60 % (when compared to patients with the low on ASA platelet activity), we had to study 88 and 176 subjects, respectively, to be able to reject the null hypothesis that the minor allele frequency for subjects with high and low on ASA platelet activity are equal with probability (power) 0.85. The Type I error probability associated with this test of this null hypothesis is 0.002 (nominal alpha level 0.05 corrected by Bonferroni method for 23 simultaneously analyzed polymorphisms). The recorded clinical data, when normally distributed in the analyzed group of patients, are presented as mean and SD, and non-normally distributed data are presented as medians and interquatrile (IQR) range. The extent to which observed genotype frequencies for each SNP deviated from the expected, under the Hardy–Weinberg equilibrium, was assessed with a Fisher exact test (p < 0.001 threshold for deviation). Individual SNP associations with high on ASA platelet activity were estimated using logistic regression for additive, recessive and dominant genetic models. Corresponding odds ratios (OR) and 95 % confidence intervals (CI) were subsequently calculated for each SNP according to the role of the SNP’s minor allele in each genetic model. All statistical analyses were performed using IBM-SPSS ver. 19 and Stata (Stata Corporation, College Station, TX) software.
Results
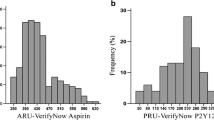
From the initially enrolled 304 patients, complete clinical data and blood samples finally became available for 297 patients. Subsequently, 8 patients were eliminated from further analysis based on the suspected ASA non-compliance (i.e., S-TXB2 concentrations >7.2 ng/ml). The remaining 289 patients were divided into 2 groups, one group (N = 202) with not high platelet reactivity on ASA based on CEPI-CT ≥ 193 s and second group (N = 87) with high platelet reactivity based on CEPI-CT < 193 s. Demographic characteristics, clinical data, and results of platelet activation and aggregation studies for the study groups are summarized in Table 1. The statistically significant differences between the study groups were recorded for all platelet reactivity studies (i.e., CEPI-CT, CADP-CT and ARU for VerifyNow Aspirin Assay), plasma von Willebrand factor (vWF) level, as well as for the frequency of coronary artery disease (CAD) and previous MIs occurrence (all higher in the CEPI-CT < 193 s group).
The total of 200,251 SNPs was finally obtained from the microarray genomic scan (Fig. 1, see also On-line Supplement for details of the selection process). The subsequent VEGAS analysis resulted in the ranking list of the selected SNPs according to the test probability in the total of 16917 genes. We have chosen for further verification 25 genes (Supplemental Table 1) and their corresponding SNPs with most significant results based on their relative ranking number, probability of being involved in the coagulation or platelet adhesion mechanisms, as well as having at least 4 SNPs analyzed within the gene boundaries. The results of individual genotyping verification are presented in Supplemental Table 1. The individual genotyping resulted in the data for 23 SNPs. Two SNPs (rs7824350, rs7331142) failed genotyping threshold because they were genotyped in <85 % of study participants. The most statistically significant results corrected for the multiple comparison with Bonferroni correction (p < 0.05/23 ~ 0.0021) were obtained for the intronic SNP rs2502448 located within the gene RGS7 on chromosome 1 (Fig. 2). Six other SNPs in various genes including DPP6 (rs1387180, p = 0.003), GSR (rs3779647, p = 0.006), CACNA1A (rs8103699, p = 0.007), ERBB4 (rs10195573, p = 0.017), ANK3 (rs2393602, p = 0.023), and AK5 (rs4949803, p = 0.044) showed nominal statically significant differences (p < 0.05 uncorrected for multiple-comparison testing) in allelic frequency between two investigated groups.

Manhattan plot of p values for differences in the pooled allelic frequency between analyzed groups against chromosomal location for all analyzed SNPs (after data filtration described in materials and methods). Horizontal axis–chromosome numbers. Probability data shown as–log of calculated p value (Minlog) on vertical axis

Identification of SNPs located in the RGS7 gene.(Upper panel) Plot of combined–log p values for allelic frequency difference (vertical axis) versus position on chromosome 1 (in kb, horizontal axis) for SNPs analyzed by the Illumina Human Omni 2.5 microarray within the locus of RGS7 gene. (Lower panel) SNPs in linkage disequilibrium (according to HapMap Release 27) with rs2502448 (measured as R2 on left vertical axis) in relation to the position on chromosome 1 (expressed in kb on horizontal axis), and approximate position of the RGS7 sequence (green arrow). The right side vertical axis indicates the recombination rate (in cM/Mb) along the nucleotide sequence on chromosome 1 fragment surrounding rs2502448
In order to further characterize the character of interactions between investigated SNPs genotypes and platelet reactivity we performed multiple additional statistical tests listed in Supplemental Table 2. Logistic regression statistics was used to determine which genetic model (recessive vs dominant vs additive) would describe best the individual genotyping data. This analysis was performed for three predetermined cut-off points of CEPI-CT assay: 165, 193 and 300 s. Normal platelet reactivity on ASA therapy was defined at several predetermined cut-off thresholds (i.e., CT = 300, 193, 165 s for CEPI-CT) and high on ASA reactivity defined as any CEPI-CT PFA-100 lower than the predefined cut-off value. The highest statistical significance (p value between 0.0001–0.008) was again observed (at all three analyzed cut-off times in CEPI-CT) for RGS7 (rs2502448) using recessive genetic model. The statistical significance using logistic regression statistics for DPP6 (rs1387180) was also observed in CEPI-CT assay for 193 s (p = 0.001, additive model) and 165 s (p = 0.0001, dominant) cut-off CTs. Another SNP, GRS (rs3779647) was also found significant in this analysis (p = 0.002, dominant model) for single CEPI-CT of 193 s. Moreover several other SNPs displayed nominal statistical significance in this model including FOSL1, CACNA1A, PLA2G4E, AK5, KCNV2, ANK3, MMP16, ERBB4 and HS6ST1 (Supplemental Table 2).
By creating the binary outcome (normal and high on ASA platelet reactivity), both OR and CI were also calculated by logistic regression for top three most significant SNPs. The results of these calculations are shown in Table 2. In the most statistically significant recessive model, carriers of both variant (minor) alleles of RGS7 (rs2502448) were characterized by increased OR of 3.45 (CI: 1.82–6.53) (for CEPI-CT < 193 threshold for high on ASA platelet reactivity when compared with carriers of at least one major wild-type).
Because of the CEPI-CT data were censored (i.e. maximum CT set at 300 s) we used censored regression (Tobit) analysis to investigate the relation between SNP genotypes and CEPI-CT data. The obtained results (Supplemental Table 2) indicate that the highest statistical significance in this model was observed for RGS7 (rs2502448, p = 0.001) and ANK3 (rs2393602, p = 0.002). In addition, several other SNPs displayed nominal statistical significance (p < 0.05, uncorrected for multiple comparisons), e.g. DPP6, MMP16, ERBB4, DPP6, KCNV2, AK5, GRS, CACNA1A, and HS6ST1. The censored regression model was also used to adjust the statistically significant results obtained in the previous stage for confounding due to significant factors identified in the demographic analysis of the cohort. The only significant covariate found in the censored regression model was the concentration of vWF (p = 0.001). Because vWF is known to affect the PFA-100 assay [13], this covariate was added to the censored regression model for each significant SNP identified in the first stage of statistical analysis. The results indicate that RGS7 (rs2502448) remained statistically significant, after correction for single significant confounding factor, as well as other significant SNPs.
No regression analysis was performed for nominally significant SNPs associated with VerifyNow Aspirin Assay and CADP-CT PFA-100 methods because the statistical significance for these SNPs could not be confirmed after multiple comparison correction.
Discussion
The present study identified several novel, putative genomic targets, which might be associated with observed inter-individual differences in the platelet reactivity CEPI-CT phenotype in diabetic patients on ASA therapy. The current study represents, to our knowledge, first GWAS and DNA pooling approach in relation to high on ASA platelet reactivity phenotype in the diabetic population during chronic ASA therapy. The allocation of the patients into one of the two study groups was decided arbitrary on the basis of their platelet activation measured by CEPI-CT at 193 s threshold. This selection was based on the results from our previous investigations indicating that CEPI-CT method proved to be most suitable for detecting genomic polymorphism in the platelet reactivity in T2DM patients during ASA treatment [3]. Our individual genotyping results show that platelet reactivity measured with CEPI-CT, but not with CADP-CT or VerifyNow, was significantly affected by different SNPs in the diabetic population treated with ASA. This finding should not be surprising, because the SNPs were selected from GWAS scan based on the differences in the platelet reactivity assessed by the CEPI-CT assay. In addition, it has been previously hypothesized, that by removing variability in platelet function associated with the thromboxane pathway, ASA treatment may increase the ability to detect the relationship between genotype and phenotype related to the alternative platelet activation pathways [14].
The most significant results were obtained for rs2502448 located in the intronic part of RGS7 gene on chromosome 1. This finding may indicate that this particular polymorphism (or other polymorphisms in LD) may be associated with the high on ASA platelet reactivity phenotype in diabetics. The magnitude of the observed effect for this polymorphism (as measured by its OR of 3.45 (CI: 1.82–6.53) using recessive genetic model) was significantly larger than for any variants investigated before using CEPI-CT method in our previous study using the same cohort of diabetic patients [3].
RGS proteins serve as guanosine triphosphatase (GTP-ase)–activating proteins for Gα and thus accelerate transmembrane signal transmission [15]. Alterations in the RGS7 gene might potentially produce functional changes in cell membrane receptors for leukotrienes, prostaglandins, lipoxins, and thromboxane receptors. Other RGS7 genetic variants have been examined previously for association with coagulation abnormalities. Among Blacks, another intronic SNP in RGS7 rs670659 was related to venous thrombosis [16]. In addition, an association between rs670659 in RGS7 and venous thrombosis in Caucasian population was also reported [17].
As many as 10 RGS proteins (in particular RGS10 and 18) are expressed in platelets, although many of them solely at the level of RNA transcripts [6–10, 18–22]. It was suggested that the role of RGS18 is to limit platelet activation [20, 23, 24]. In GWAS study conducted by Johnson et al. [25] in healthy subjects, SNPs in RGS18 were found to be associated with both epinephrine- and ADP-induced platelets aggregation. It is important to note that RGS18 has approximately 30–40 % identity to other RGS proteins [26]. Less data is available for the RGS7 protein, which is expressed in brain tissue, but also in platelets [21]. It has been hypothesized that RGS7 could influence physiological factors associated with platelet function through central modulation of serotonin 2A (5-HT2A) receptors and α2-adrenoreceptors [27, 28]. These receptors have been observed as expressed in the periphery on vascular muscle cells and platelets among others and their function includes vasoconstriction and platelet aggregation. It was suggested that antidepressant olanzapine-induced activation of the JAK-STAT signaling cascade causes increased expression of RGS7, which in turn could mediate desensitization of 5-HT2A receptor signaling caused by olanzapine since RGS7 binds to Gαq protein and accelerates GTP hydrolysis. This action could in part explain the inhibitory action of olanzapine on platelet aggregation induced by ADP.
The RGS7 SNP (rs2502448) is located within currently assumed noncoding, intronic sequence. There are several potential mechanisms that could explain its association with the platelet response and warrant further study. For example, this SNP may be in LD with promoter SNPs that have not yet been identified or that were not genotyped in this study. Furthermore, this intronic SNP may potentially affect receptor function through alternative splicing mechanisms. In addition to the association with RGS7 SNP (rs2502448), we also found an association of CEPI-CT assay with intronic SNP (rs1387180) in the DDP6 gene that encodes dipeptidyl aminopeptidase X (DPPX). Product of this gene binds specific voltage-gated potassium channels and alters their expression and biophysical properties [29]. We also found another significant intronic SNP (rs3779647) in the GSR gene. This gene codes for the glutathione reductase, which is involved in preventing the accumulation of hydroperoxides and plays a role in formation of arachidonic acid metabolites [30]. In addition, we identified eight other SNPs: (CACNA1A-rs8103699, ERBB4-rs10195573, ANK3-rs2393602, AK5-rs494803, MMP16-rs2664369, KCNV2-rs1006698, HS6ST1-rs6711447, FOSL1-rs10791830) that were potentially associated with inter-individual differences in the platelet activation measured with PFA-100 assay in the cohort of diabetic patients on ASA treatment. All of them had not been studied previously, and all association signals were novel.
To date small numbers of human GWAS studies have been conducted for platelet function. In most of these studies platelet reactivity was measured using both epinephrine- and/or ADP-induced aggregation in platelet-rich plasma (PRP). Mathias et al. identified several potential loci associated with platelet aggregation (e.g., MME-membrane metalloendopeptidase, on chromosome 3q21-27, associated with ASA response to collagen induced aggregation in PRP [14]. In addition, Johnson et al. identified other genomic regions strongly associated with PRP ADP (PEAR1, MRVI1, SHH), PRP epinephrine (ADRA2A, PEAR1, PIK3CG, JMJD1C) and collagen (GP6) responses in two European ancestry cohorts with replication in one African ancestry cohort [25]. Guerrero et al. [31] in a GWAS study on 75 children found a relation between increased platelet reactivity determined by light aggregation and LPAR1 and MYO5B genes that was subsequently confirmed in the replication samples. These data, along with our results, suggest that relatively large number of genetic polymorphisms may have functional effect on platelet reactivity, and its detection may depend on studied population and methods used for determination of platelet reactivity.
Study limitations
The results obtained in this GWAS should be interpreted with caution because of the lack of control group without diabetes, which would allow to directly assessing the impact of diabetes and ASA treatment on measured phenotype (i.e. platelet reactivity) and its association with new SNPs. Another limitation is related to the choice of tests for measurement of platelet activation. Light transmission aggregometry is considered to be the gold standard for platelet function test but is poorly standardized, requires a specialist laboratory, and is unlikely to be used widely in routine clinical practice [32]. Thus, in our study we assessed platelet reactivity by using three point-of-care tests (CEPI-CT and CADP-CT by PFA-100, and ARU by VerifyNow Aspirin Assay) that are being used in multiple centers instead of the “gold standard” LTA. Other weaknesses of our study include limited sample size for linkage and the lack of external replication in independent cohort of diabetic patients. Lastly, most of our associations might be either random or associated with the particular patient population (e.g. diabetes), and this question could not be answered in this study. Replication studies are needed to determine if any of the results we report are indicative of true associations.
Conclusions
In summary, the untargeted genome-wide approach to detect genetic associations of platelet reactivity during ASA treatment in diabetic patients provides an opportunity to identify putative, novel biologic pathways related to platelet activation in the diabetic population and to direct future studies of candidate genes that hold the most promise for relevance to platelet activation. Enhancing our understanding of the mechanisms responsible for platelet activation in diabetic patients may in turn identify novel strategies for antiplatelet therapy in this specific population. We believe that the polymorphisms in these genes may be of clinical importance. Further studies in larger diabetic and non-diabetic populations are needed to confirm and extend the results we observed in this relatively small, but ethnically homogenous, cohort.
References
Mortensen SB, Larsen SB, Grove EL et al (2010) Reduced platelet response to aspirin in patients with coronary artery disease and type 2 diabetes mellitus. Thromb Res 126:e318–e322
Davi G, Patrono C (2007) Platelet activation and atherothrombosis. N Engl J Med 357:2482–2494
Postula M, Kaplon-Cieslicka A, Rosiak M et al (2011) Genetic determinants of platelet reactivity during acetylsalicylic acid therapy in diabetic patients: evaluation of 27 polymorphisms within candidate genes. J Thromb Haemost 9:2291–2301
Nielsen HL, Kristensen SD, Thygesen SS et al (2008) Aspirin response evaluated by the VerifyNow aspirin system and light transmission aggregometry. Thromb Res 123:267–273
Kunicki TJ, Williams SA, Salomon DR et al (2009) Genetics of platelet reactivity in normal, healthy individuals. J Thromb Haemost 7:2116–2122
Chakroun T, Addad F, Abderazek F et al (2007) Screening for aspirin resistance in stable coronary artery patients by three different tests. Thromb Res 121:413–418
Gonzalez-Conejero R, Rivera J, Corral J et al (2005) Biological assessment of aspirin efficacy on healthy individuals: heterogeneous response or aspirin failure? Stroke 36:276–280
Janicki PK, Vealey R, Liu J et al (2011) Genome-wide association study using pooled DNA to identify candidate markers mediating susceptibility to postoperative nausea and vomiting. Anesthesiology 115:54–64
Macgregor S, Zhao ZZ, Henders A et al (2008) Highly cost-efficient genome-wide association studies using DNA pools and dense SNP arrays. Nucleic Acids Res 36:e35
Liu JZ, McRae AF, Nyholt DR et al (2010) A versatile gene-based test for genome-wide association studies. Am J Hum Genet 87:139–145
Skol AD, Scott LJ, Abecasis GR et al (2006) Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38:209–213
Dupont WD, Plummer WD (1990) Power and sample size calculations. A review and computer program. Control Clin Trials 11:116–128
Haubelt H, Anders C, Vogt A et al (2005) Variables influencing Platelet Function Analyzer-100 closure times in healthy individuals. Br J Haematol 130:759–767
Mathias RA, Kim Y, Sung H et al (2010) A combined genome-wide linkage and association approach to find susceptibility loci for platelet function phenotypes in European American and African American families with coronary artery disease. BMC Med Genomics 3:22
De Vries L, Gist Farquhar M (1999) RGS proteins: more than just GAPs for heterotrimeric G proteins. Trends Cell Biol 9:138–144
Austin H, De Staercke C, Lally C et al (2011) New gene variants associated with venous thrombosis: a replication study in White and Black Americans. J Thromb Haemost 9:489–495
Bezemer ID, Bare LA, Doggen CJ et al (2008) Gene variants associated with deep vein thrombosis. JAMA 299:1306–1314
Pearson JV, Huentelman MJ, Halperin RF et al (2007) Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am J Hum Genet 80:126–139
Signarvic RS, Cierniewska A, Stalker TJ et al (2010) RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood 116:6092–6100
Gegenbauer K, Elia G, Blanco-Fernandez A et al (2012) Regulator of G-protein signaling 18 integrates activating and inhibitory signaling in platelets. Blood 119:3799–3807
Rowley JW, Oler AJ, Tolley ND et al (2011) Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood 118:e101–e111
Ma P, Cierniewska A, Signarvic R et al (2012) A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood 119:1935–1945
Kim SD, Sung HJ, Park SK et al (2006) The expression patterns of RGS transcripts in platelets. Platelets 17:493–497
Garcia A, Prabhakar S, Hughan S et al (2004) Differential proteome analysis of TRAP-activated platelets: involvement of DOK-2 and phosphorylation of RGS proteins. Blood 103:2088–2095
Johnson AD, Yanek LR, Chen MH et al (2010) Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet 42:608–613
Gagnon AW, Murray DL, Leadley RJ (2002) Cloning and characterization of a novel regulator of G protein signalling in human platelets. Cell Signal 14:595–606
Jedema HP, Gold SJ, Gonzalez-Burgos G et al (2008) Chronic cold exposure increases RGS7 expression and decreases alpha(2)-autoreceptor-mediated inhibition of noradrenergic locus coeruleus neurons. Eur J Neurosci 27:2433–2443
Shi J, Damjanoska KJ, Zemaitaitis B et al (2006) Alterations in 5-HT2A receptor signaling in male and female transgenic rats over-expressing either Gq or RGS-insensitive Gq protein. Neuropharmacology 51:524–535
Niwa N, Nerbonne JM (2010) Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol 48:12–25
Hill TD, White JG, Rao GH (1989) Role of glutathione and glutathione peroxidase in human platelet arachidonic acid metabolism. Prostaglandins 38:21–32
Guerrero JA, Rivera J, Quiroga T et al (2011) Novel loci involved in platelet function and platelet count identified by a genome-wide study performed in children. Haematologica 96:1335–1343
Michelson AD (2009) Methods for the measurement of platelet function. Am J Cardiol 103:20A–26A
Acknowledgments
Dr Postula was supported by a Fulbright Fellowship from USA State Department. The AVOCADO study was supported financially as part of the research grant from the Polish Pharmaceutical Company ADAMED for a Young Scientist 2007 Award [grant number: 1WR DAR1/2007] and Polish Cardiac Society Club 30 grant 2010. The authors thank Thutrang Nguyen, B.A. from the SNP Genotyping Core Facility at Children’s Hospital Boston, Massachusetts, for performing the custom Sequenom iPLEX assay.
Disclosure of conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Marek Postula and Piotr K. Janicki contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Postula, M., Janicki, P.K., Rosiak, M. et al. New single nucleotide polymorphisms associated with differences in platelets reactivity in patients with type 2 diabetes treated with acetylsalicylic acid: genome-wide association approach and pooled DNA strategy. J Thromb Thrombolysis 36, 65–73 (2013). https://doi.org/10.1007/s11239-012-0823-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-012-0823-6