
Abstract
In recent years, hypoxic cell radiosensitizers have evolved as potential molecules in the diagnosis of cancer and in clinical radiotherapy. Nitroimidazole and its sulfonamide analogues are effective radiosensitizers working on hypoxic tumor cells. The application of QSAR modeling technique has paved an easier way for the prediction of newly developed compounds. In the present study, we have used 21 nitroimidazole sulfonamide analogues to develop 2D quantitative structure-activity relationship (QSAR) models and determine their structural features essential for two radiosensitization properties, viz., sensitizer enhancement ratio and survival ratio. The models were developed using the small dataset modeler software (http://teqip.jdvu.ac.in/QSAR_Tools/DTCLab/), and model validation was performed using various stringent validation criteria. The developed models are robust, predictive, and should be useful tools to predict the radiosensitization of nitroimidazole sulfonamides. Furthermore, we have used the “prediction reliability indicator” tool to check the predictive ability of the developed models using 14 external nitroimidazole sulfonamide derivatives. We have also developed quantitative structure-activity-activity relationship (QSAAR) models for the two endpoints.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypoxia is a principal component of the tumor microenvironment, which is considered to be the pivotal cause of clinical radioresistance and local failure. Oxygen is considered as the best radiosensitizer by far; however, metabolic consumption of oxygen limits its diffusion into hypoxic tumor cells [1]. Hypoxia has a chief role in cancer progression manipulating angiogenesis [2], vasculogenesis [3], and activation of a glycolytic shift in metabolism [4], invasion enhancement, and metastasis [5]. Radiation therapy is an anchoring treatment for many types of cancer; however, there is a great challenge to augment radiation damage to the tumor tissues and reduce side effects to healthy tissues. Radiosensitizers are promising agents in controlling hypoxia by enhancing tumor tissue injury through accelerating DNA damage and producing free radicals [6].
Oxygen-mimetic radiosensitizers are potential agents in controlling radiation damage in hypoxic tumor cells. Nitroheterocyclic compounds such as nitroimidazoles have been evaluated as oxygen-mimetic agents where electron-rich nitro group is intended to react with DNA radicals produced by ionizing radiation in a similar fashion like oxygen does [6, 7]. DNA and nitro group adduct leads to DNA strand breaks and subsequent cellular apoptosis or lysis. Enhanced radiosensitization after prolonged exposure of cells to misonidazole was identified by Hall et al. [8]. However, this was restricted by delayed peripheral neuropathies when combined with fractionated radiotherapy [9]. Ro 03-8799 (pimonidazole) and SR 2508 (etanidazole) were used in combination as cell radiosensitizers in the treatment of high-grade gliomas. It was found that Ro 03-8799 is distributed extensively in the central nervous system, and SR 2508 could achieve high tumor concentrations when the blood-brain barrier is compromised [10]. Yahiro et al. studied effects of the radiosensitizer doranidazole (PR-350) on the radioresponse of murine and human tumor cells in vitro and in vivo and observed that the amount of radiosensitization of tumors induced by doranidazole is dependent on the oxygenation status of the tumors [11]. A 5-nitroimidazole derivative, nimorazole, has shown similar radiosensitization properties to misonidazole at clinically acceptable dose levels. It is clinically used in head and neck cancer along with fractionated radiotherapy (FRT) [12].
Recently, a wide range of nitroimidazole sulfonamides has been identified as potential radiosensitizers against hypoxic cancer cells [13, 14]. These sulfonamides have been considered as hypoxia-selective cytotoxins and radiosensitizers, and their variation in side chains noticeably influence the physicochemical properties of the analogues. The compounds might have lowered aqueous solubility and raised the electron affinity of the nitroimidazole group.
Computational approaches such as quantitative structure-activity/property relationships (QSAR/QSPR) [15] are effective tools in prediction of radiosensitization properties when experimental data is scarce. The method allows virtual screening of drug libraries to find suitable drug-target for a particular disease. QSAR finds an immense application in the prediction of ADMET (absorption, distribution, metabolism, elimination, and toxicity) properties of drugs and other biologicals [16, 17]. A large number of researches have been carried out with the hope to do some predictions of the ADMET properties using the structural features of the molecules. QSAR/QSPR modeling is one such important approach where data derived from their activity profiles and their different structural features (quantitative molecular descriptors) are used [18]. Radiosensitization is a property of nitroimidazole and nitroimidazole sulfonamide derivatives and can thus be subjected to QSAR analysis. A well-validated QSAR model could evaluate and generate radiosensitization data for such related compounds when experimental data is not available.
The present study explores the features essential to show radiosensitization properties by nitroimidazole sulfonamide derivatives using QSAR and quantitative structure activity-activity relationship (QSAAR) modeling [19]. Two dimensional (2D) descriptors obtained from Dragon and SiRMS software were utilized during the development of well-validated models. A small dataset of nitroimidazole sulfonamides is used for modeling in the current study where splitting of the dataset into training and test sets would cause loss of chemical information leading to unreliable models. Thus, a “small dataset modeling” approach has been adopted using the whole dataset [20], and the developed models were subjected to leave-many-out cross-validation. Furthermore, a group of nitroimidazole sulfonamides has been predicted to prove the validity of the developed models.
Materials and methods
Dataset
In vitro radiosensitization data of selected compounds involving sensitizer enhancement ratio (drug SER) and survival ratio (drug SR) was obtained from a previously published research work [21]. A dataset of 21 compounds given in Table 1 was selected for 2D QSAR modeling. Sensitizer enhancement ratio (SER) can be defined as the ratio of radiation dose for 1% survival without or with the drug in a condition where HCT116 cells (human colorectal carcinoma cell line) were exposed to the drug at 6–29 Gy radiation for 1 h. Survival ratio can be explained using the following expression: “SR= (cell survival with radiation)/(cell survival with drug and with radiation) interpolated from the radiation dose response curves at 15 Gy.” During modeling, the drug SER values were used as provided in the original article but drug SR values were converted into their logarithmic form (logSR) for analysis. The compounds were drawn in MarvinSketch software (version 14.10.27) [22] with hydrogen bond addition and proper aromatization and saved as MDL.mol, a suggested format for further descriptor calculation.
Molecular descriptors
The molecular descriptor is the “final result of a logical and mathematical procedure, which transforms chemical information encoded within a symbolic representation of a molecule into a useful number or the result of some standardized experiment” [23]. A selected class of 356 2D molecular descriptors was calculated from Dragon version 7 [24] software. These comprised E-state indices, connectivity, constitutional, functional, 2D atom pairs, ring, atom-centered fragments, and molecular property descriptors. Intercorrelated (|r| > 0.95) and constant (variance < 0.0001) variables and other incompetent data were removed using a software available at http://dtclab.webs.com/software-tools prior to model development. This resulted in 224 Dragon descriptors which were used for modeling. Furthermore, SiRMS descriptors were calculated using SiRMS (version 4.1.2.270) [25] tool and used along with Dragon descriptors during modeling. Simplex representations of molecular structure (SiRMS) descriptors are a class of molecular descriptors developed from 1D to 4D molecular structures involving tetratomic fragments of different simplex descriptors having predefined chirality, composition, and symmetry [25].
Model development: application of small dataset modeler
Before development of a QSAR model, the dataset is generally divided into a training set (calibration) and a test set (validation). Furthermore, a double cross-validation method [26] of model development involves two nested cross-validation loops: internal (inner) and external (outer) cross-validation loops. In the outer loop, the data points are segregated into two subsets, i.e., training and test sets. The training set is further employed in the inner loop for model building and selection purpose. The test set has the sole purpose of model validation. However, the present study deals with a small dataset containing a limited number of data points (21 compounds), and splitting of this dataset into training and test sets is not desirable. Small dataset modeling (http://teqip.jdvu.ac.in/QSAR_Tools/DTCLab/) involves the DCV method of modeling for small datasets without dividing the dataset into training and test sets [20]. Here, the “modeling set” in the inner loop is not generated. However, deriving all possible combinations (k) of the validation set (containing n compounds) and the calibration set (containing n − r compounds) is followed. The tool has an option for the user to define the number of compounds to be kept in the validation set (r) depending on which the calibration and validation sets are defined. Calibration set compounds are used for the generation of genetic algorithm-multiple linear regression (GA-MLR) [27, 28] models, and the validation sets are utilized for model prediction purpose. A number of internal and external validation metrics are calculated in the exhaustive double cross-validation technique for all the selected models. Additionally, the software also derives partial least squares (PLS) [29] regression models corresponding to each MLR model. Furthermore, the selection of best/top model can be done in any of the five following methods mentioned:
-
(i)
Model (MLR/PLS) with the lowest mean absolute error or MAE (95%) in the validation set is selected.
-
(ii)
Model (MLR/PLS) with the lowest MAE (95%) in the modeling set is selected.
-
(iii)
Model (MLR/PLS) with the highest \( {Q}_{\mathrm{Leave}-\mathrm{many}-\mathrm{out}}^2 \) (modeling set).
-
(iv)
Application of consensus modeling by using top ranking models selected based on the MAE (95%) values in the respective validation sets. Two types of consensus approaches include (a) simple arithmetic average of predictions from all the selected top models, and (b) weighted average of predictions by assigning appropriate weights to the selected top models based on the mean absolute error obtained from leave-one-out cross-validation, MAEcv(95%).
-
(v)
A pool of unique descriptors from the top 3 models with lowest MAE (95%) of the validation set is used. These descriptors are used for further model development purpose. In case of MLR, the best subset selection (BSS) method is used which finds the best combinations of descriptors out of all the possible combinations of unique descriptors present in the selected models. In case of PLS models, the models are formed by all descriptors selected in the top models through a PLS run.
The approach proposed in small dataset modeler (Fig. 1) thus ensures the division of small dataset internally within the DCV algorithm without the actual need of a test set. Thus, there is no requirement of the dataset division. The small dataset modeling approach combines data curation, exhaustive double cross-validation, and optimal model approaches including consensus predictions for model development, particularly for small datasets.

The approach adopted to develop QSAR models for small-sized dataset using small dataset modeler
Statistical validation metrics
A rigorous analysis using multiple approaches of assessment of the model quality for measurement of the fitness, stability, robustness, and predictivity of the developed models was carried out. In the present work, we have computed various statistical parameters like determination coefficient (R2) and leave-one-out squared correlation coefficient (\( {Q}_{\mathrm{LOO}}^2 \)) for internal validation. We have also calculated the leave-many-out squared correlation coefficient (\( {Q}_{\mathrm{LMO}\left(20\%\right)}^2 \)) for the final PLS models [30]. Furthermore, \( {r}_m^2 \) metrics [31], root mean square error (RMSE), and mean absolute error (MAE) were also calculated [32].
Results and discussion
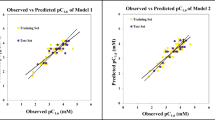
2D QSAR models using Dragon and SiRMS descriptors explaining chemical features required for good drug radiosensitization (both SER and logSR) are shown in the following section. There are 4 models developed of which two are QSAR models and the rest two are QSAAR models. All the models are three-descriptor PLS models with 2 latent variables (LVs) showing acceptable values for all validation metrics as shown in Table 2. The validation metrics included R2, Q2, \( {Q}_{\mathrm{LMO}\ \left(20\%\right)}^2 \), \( \overline{r_{m\left(\mathrm{LOO}\right)}^2} \), \( {\varDelta r}_{m\left(\mathrm{LOO}\right)}^2 \), SD (95% data; training), MAE (95% data; training), and RMSE. Furthermore, we have calculated the \( {Q}_{F1}^2 \) metric for the validation set in each iteration cycle for each model during the calculation of \( {Q}_{\mathrm{LMO}\ \left(20\%\right)}^2 \) (Supplementary Section). The experimental and predicted values for all the models are given in Supplementary files (S1) and the observed versus predicted plots for all the developed QSAR and QSAAR models are shown in Fig. 2. The different PLS plots including variable importance plot [33], loading plot [29], regression coefficient plot [29], and randomization plot [34] discussed later are shown in Supplementary files (SM2).

Scatter plots for QSAR and QSAAR models
Model 1: modeling drug sensitizer enhancement ratio
The first descriptor H-049 belongs to atom-centered fragment type, which indicates H atom attached to C3 (sp3)/C2 (sp2)/C3 (sp2)/C3 (sp). The descriptor symbolizes the hydrogen of a CH group with the carbon bonded to varying numbers of heteroatoms in a variety of hybridizations. The descriptor has a positive contribution towards the response (Fig. 3) which is well understood from certain higher active compounds in the dataset like compounds 19 (SER = 1.81) and 24 (SER = 1.81), each of which has two H-049 fragments. On the other hand, compounds like 12 (SER = 1.11) and 16 (SER = 1.105) having only one such fragments have low SER values.

Features increasing or decreasing SER values as explained in model 1
The next descriptor is B05[O-S], which is a 2D atom pair descriptor demonstrating the presence or absence of oxygen and sulfur atoms at the topological distance 5. The negative contribution explains that presence of oxygen and sulfur atoms at the topological distance 5 will lower the SER values (Fig. 3) as observed in compounds 7 (SER = 1.11) and 16 (SER = 1.105). On the other hand, in compounds like 4 (SER = 1.835) and 30 (SER = 1.687), the absence of such fragment does not lower the SER value.
The descriptor F05[C-S], another 2D atom pair descriptor, denotes the frequency of C-S at the topological distance 5. The positive contribution of the descriptor indicates that higher frequency of the C-S fragment at the topological distance 5 will increase the SER value (Fig. 3) as seen in compounds 30 (F05[C-S] = 3, SER = 1.68) and 38 (F05[C-S] = 3, SER = 1.67).
Model 2: modeling drug survival ratio (logSR)
S_A(chg)/A_B_B_D/1_4s,3_4s/4 represents a four atomic fragment labeled by partial charges, and its negative regression coefficient indicates that it reduces the radiosensitization property with the presence of such fragment (shown in Fig. 4). In compounds like 26 and 28, presence of such fragment reduces the radiosensitization (logSR = 0.681 and 0.208).

Factors decreasing logSR values as explained in model 2
C-033 is an atom-centered fragment descriptor represented by R--CH..X fragment. “R” denotes any group linked through carbon, “- -“ represents an aromatic bond as in benzene or delocalized bonds such as the N-O bond in a nitro group, “..” represents aromatic single bonds as the C-N bond in pyrrole, and “X” is any electronegative atom (O, N, S, P, Se, halogens) [35]. The negative coefficient indicates that presence of this type of fragment lowers logSR (Fig. 4) values as observed in compounds 6 (C-033 = 1, logSR = 0.462) and 7 (C-033 = 1, logSR = 0.255).
F07[C-C] is a 2D atom pair descriptor, which signifies the frequency of the C-C fragment at the topological distance 7. The negative coefficient indicates that a higher value of the descriptor may decrease the radiosensitization (logSR value) (Fig. 4). This is observed in compounds like 12 and 8 where F07[C-C] are high (6 and 5 respectively) and their logSR values are low (0.301 and 0.591 respectively).
Quantitative structure activity-activity relationship models
Quantitative structure activity-activity relationship (QSAAR) models are mathematical expressions correlating two biological endpoints, here SER and logSR, with the aim to extrapolate any one explicit activity endpoint when the experimental data is not available. This advanced technique can overcome the additional cost of manifold experimental procedures. In the present study, we have developed two QSAAR models, one taking SER as the endpoint and logSR as an independent variable and another taking logSR as the endpoint and SER as an independent variable. It was found that these two endpoints had positive correlation between themselves explaining that increase in experimental values of any of the endpoints would increase the other endpoint values and vice versa.
Model 3: QSAAR modeling of SER
Model 3 is a PLS model with 2 latent variables and shows acceptable values of the validation metrics. Here, logSR has been used as an independent variable to produce a QSAAR model for drug SER. Thus, for any compound, if survival ratio (SR) value is known, the SER value can be extrapolated using model 3. This reduces time and experimental expenses. In the model, logSR shows a positive regression coefficient; hence, a higher value of logSR will increase SER values as observed in compounds like 19 (logSR = 2.212, SER = 1.81) and 24 (logSR = 2.057, SER = 1.81).
The descriptor F03[C-C] is a 2D atom pair descriptor signifying the frequency of C-C fragments at the topological distance 3. This makes a positive contribution to the endpoint, thus indicating that with an increase in the F03[C-C] descriptor value, SER value will also increase as seen in compounds 30 (F03[C-C] = 14, SER = 1.68) and 35 (F03[C-C] = 13, SER = 1.71). Another 2D atom pair descriptor T(N..O) appears in the model signifying the sum of topological distances between N..O. This descriptor has a negative influence on the SER values indicating that the total distance between nitrogen and oxygen should be low for higher SER values as in compound 4 (T(N..O) = 51, SER = 1.8). Compounds with higher T(N..O) values will have lower SER values as observed in compounds 8 (T(N..O) = 130, SER = 1.28) and 12 (T(N..O) = 106, SER = 1.11). Features increasing and decreasing SER values are shown in Fig. 5.

Features increasing or decreasing SER value as explained in model 3
Model 4: QSAAR modeling of logSR
In model 4, SER has been used as an independent variable for modeling logSR. SER makes a positive contribution to logSR, proving the authenticity of the previously developed model 3 and this can be explained by the same compounds 19 and 24.
F03[C-C] is a 2D atom pair descriptor symbolizing the frequency of the C-C fragment at the topological distance 3. The descriptor shows a negative regression coefficient, thus signifying that with an increase in F03[C-C] values, logSR value will decrease and vice versa. It is observed that in compounds 15 and 34, the F03[C-C] values are high (10 and 11 respectively) and their logSR values are low (log SR = 0.699 and 1.134 respectively). The opposite is observed in compounds 19 (F03[C-C] = 2, logSR = 2.057) and 24 (F03[C-C] = 4, logSR = 2.212) having lower values for F03[C-C]. Descriptor nO is a constitutional descriptor meaning the number of oxygen atoms present in a molecule. The positive regression coefficient indicates that presence of oxygen atoms is beneficial for the in vitro radiosensitization (logSR). In compounds like 19 (logSR = 2.057) and 24 (logSR = 2.212), higher number of oxygen (nO = 5) contributes to a higher value of logSR. Features increasing and decreasing logSR value are shown in Fig. 6.

Features increasing or decreasing logSR value as explained in model 4
Plot interpretation
-
(i)
Variable importance plot (VIP)—A VIP can provide with a better knowledge about the descriptors and their contribution in controlling the radiosensitization properties of nitroimidazole sulfonamides. The plot signifies the order of contribution of each descriptor appearing in the model. The most and least important descriptors can be identified using this plot. A variable with VIP score > 1 indicates the descriptor has higher statistical significance as compared to the one with a lower VIP value [33]. The VIP plot showing the descriptors from higher to lower significance is given in the Supplementary Section S2 (Figs. S1–S4).
-
(ii)
Loading plot—The loading plot defines the relationship between X variables and Y variables [29]. The plot was developed using the two latent variables for all the four models. The plot describes the impact of the different variables. Descriptors that are grouped together have similar meanings and similar effects on the response, whereas descriptors with different meanings are situated at a considerable distance from each other. Descriptors which are situated far from the plot origin have greater impact on the response. The loading plots of the four models are given in the Supplementary Section S2 (Figs. S5–S8).
-
(iii)
Regression coefficient plot—The regression coefficient plot [29] gives knowledge about the positive or negative contribution of the descriptors towards the activity (SER or logSR) of the compounds. Descriptors having a positive regression coefficient indicate that with an increase in the descriptor values, the SER and logSR increase. On the other hand, a negative regression coefficient indicates that with an increase in the descriptor value, the SER and logSR decrease. The regression coefficient plots are given in Supplementary Section S2 (Figs. S9–S12).
-
(iv)
Randomization plot—Model randomization is done to ensure that the model is not the result of any chance correlation [34]. The statistical significance of the model is determined by a randomization model. During the model randomization, multiple models are generated by shuffling different combinations of X or Y variables (here Y variable) based on the fit of the reordered model. Here, we have used 100 permutations for each model for random model generation. A model not generated out of chance correlation should have poor statistics (\( {R}_y^2 \)intercept should not exceed 0.3 and \( {Q}_y^2 \) intercept should not exceed 0.05). The randomization plots given in Figs. S12–S16 show that the developed models are non-random and robust and are suitable for prediction.
Applicability domain
Applicability domain (AD) explains the prediction reliability of a particular model. It is the “chemical space from which a model is derived and where a prediction is considered to be reliable” [36]. AD evaluation was done using DModX (distance to model) in the X-space using SIMCA 16.0.2 software (https://landing.umetrics.com/downloads-simca). The AD plots are given in Figs. 7 and 8. It is found that there is no outlier in any of the four models developed at 95% confidence level (D-crit = 0.009999).

DModX applicability domain plot of model 1 and model 2

DModX applicability domain of model 3 and model 4
Prediction dataset
A QSAR model helps in the prediction of external datasets based on their molecular features, thereby reducing the experiment costs and animal handling. To study the predictive power of the developed models, we have used 14 compounds whose SER and logSR values have been predicted. These 14 compounds were selected from Table 1 of the source article [21]. This table contained about 36 nitroimidazole sulfonamides out of which 21 compounds were used for QSAR and QSAAR modeling and rest 14 compounds were used as an external set for prediction. Furthermore, we have analyzed the prediction quality and domain of applicability using the prediction reliability indicator tool [37]. The prediction status and domain of applicability are given in Table 3. Prediction was possible for model 1 (M1), model 2 (M2), and model 3 (M3). In M1 and M2, the predicted SER and predicted logSR values were calculated for 14 compounds. In case of M3 (QSAAR-SER), SR15 values were obtained from source article [21] and the values were converted to logarithmic form and used as an independent variable for the calculation of predicted SER values. Prediction for model M4 was not possible since experimental SER values for the prediction compounds are not available. During prediction with model M1, three compounds had bad/unreliable predictions. This is due to the difference between the mean of the training set response and predicted value of the query compound being considerably higher. However, these compounds fall inside the AD of the model. In case of M2, one compound (compound no. 25) is outside AD; however, it shows moderate prediction quality. During prediction with model M3, all the compounds are found to have “moderate” prediction quality and are inside the model AD.
Conclusion
This study aims at developing 2D QSAR models with the notion to investigate the essential features in nitroimidazole sulfonamide analogues to show radiosensitization properties with respect to sensitizer enhancement ratio and survival ratio endpoints. The different descriptors obtained give an idea about the position of the features and type of chemical groups required to enhance or hinder these properties. Moreover, QSAAR modeling helps in correlating two endpoints (SER and logSR) and suggests how to extrapolate an endpoint if the experimental information is unavailable. The current study emphasizes on the application of the “small dataset modeler” software when the dataset is small and splitting of dataset is not worthy. Furthermore, the newly developed models were used for prediction of 14 compounds and their prediction reliability was checked. These developed QSAR and QSAAR models are able to predict newly developed nitroimidazole sulfonamide derivatives with known structural features. The complete overview of the work is explained in Fig. 9.

Overview of the present work involving the development of QSAR and QSAAR model using small dataset modeler
Data availability
Raw data of the modeling study are available in Supplementary materials.
References
De Ridder M, Verellen D, Verovski V, Storme G (2008) Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide 19:164–169
Muz B, de la Puente P, Azab F, Azab AK (2015) The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 3:83
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Investig 120:694–705
Chiche J, Brahimi-Horn MC, Pouysségur J (2010) Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med 14:771–794
Hill RP, Marie-Egyptienne DT, Hedley DW (2009) Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol 19:106–111
Wardman PJCO (2007) Chemical radiosensitizers for use in radiotherapy. Clin Oncol 19:397–417
Suto MJ (1991) Radiosensitizers. Annu Rep Med Chem 26:151–160
Hall EJ, Astor M, Biaglow J, Parham JC (1982) The enhanced sensitivity of mammalian cells to killing by X rays after prolonged exposure to several nitroimidazoles. IJROBP 8:447–451
Saunders M, Dische S (1996) Clinical results of hypoxic cell radiosensitisation from hyperbaric oxygen to accelerated radiotherapy, carbogen and nicotinamide. Br J Cancer 27:S271
Newman HFV, Bleehen NM, Ward R, Workman P (1988) Hypoxic cell radiosensitizers in the treatment of high grade gliomas: a new direction using combined Ro 03-8799 (pimonidazole) and SR 2508 (etanidazole). IJROBP 15:677–684
Yahiro T, Masui S, Kubota N, Yamada K, Kobayashi A, Kishii K (2005) Effects of hypoxic cell radiosensitizer doranidazole (PR-350) on the radioresponse of murine and human tumor cells in vitro and in vivo. J Radiat Res 46:363–372
Metwally MAH, Frederiksen KD, Overgaard J (2014) Compliance and toxicity of the hypoxic radiosensitizer nimorazole in the treatment of patients with head and neck squamous cell carcinoma (HNSCC). Acta Oncol 53:654–661
Hong CR, Wang J, Hicks KO, Hay MP (2016) Efficient protocol for the identification of hypoxic cell radiosensitisers. Tumor Microenviron pp. 269–290
Bonnet M, Hong CR, Gu Y, Anderson RF, Wilson WR, Pruijn FB, Wang J, Hicks KO, Hay MP (2014) Novel nitroimidazole alkylsulfonamides as hypoxic cell radiosensitisers. Bioorg Med Chem 22:2123–2132
Roy K (2015) Quantitative structure-activity relationships in drug design, predictive toxicology, and risk assessment. IGI Global
Hansch C, Leo A, Mekapati SB, Kurup A (2004) Qsar and Adme. Bioorg Med Chem 12:3391–3400
Klein C, Kaiser D, Kopp S, Chiba P, Ecker GF (2002) Similarity based SAR (SIBAR) as tool for early ADME profiling. J Comput Aided Mol Des 16:785–793
Tareq Hassan Khan M (2010) Predictions of the ADMET properties of candidate drug molecules utilizing different QSAR/QSPR modelling approaches. Curr Drug Metab 11:285–295
Lessigiarska I, Worth AP, Netzeva TI, Dearden JC, Cronin MT (2006) Quantitative structure–activity–activity and quantitative structure–activity investigations of human and rodent toxicity. Chemosphere 65:1878–1887
Ambure P, Gajewicz-Skretna A, Cordeiro MND, Roy K (2019) New workflow for QSAR model development from small data sets: Small Dataset Curator and Small Dataset Modeler. Integration of data curation, exhaustive double cross-validation, and a set of optimal model selection techniques. J Chem Inf Model 59:4070–4076
Bonnet M, Hong CR, Wong WW, Liew LP, Shome A, Wang J, Gu Y, Stevenson RJ, Qi W, Anderson RF, Pruijn FB (2018) Next-generation hypoxic cell radiosensitizers: nitroimidazole alkylsulfonamides. J Med Chem 61:1241–1254
MarvinSketch software, https://www.chemaxon.com. Accessed on 26 Nov 2020
Todeschini R, Consonni V (2009) Molecular descriptors for chemoinformatics. Wiley-VCH, Weinheim
Dragon version 7, Kodesrl, Milan, Italy, 2016; software available at http://www.talete.mi.it/index.htm. Accessed on 28 Nov 2020
Kuz’min VE, Artemenko AG, Polischuk PG, Muratov EN, Hromov AI, Liahovskiy AV, Andronati SA, Makan SY (2005) Hierarchic system of QSAR models (1D–4D) on the base of simplex representation of molecular structure. J Mol Model 11:457–467
Roy K, Ambure P (2016) The “double cross-validation” software tool forMLR QSAR model development. Chemom Intell Lab Syst 159:108–126
Devillers J (1996) Genetic algorithms in molecular modeling. Academic Press, Cornwall, Great Britain
Venkatasubramanian V, Sundaram A (2002) Genetic algorithms: introduction and applications. Encycl Comput Chem 2
Wold S, Sjöström M, Eriksson L (2001) PLS-regression: a basic tool of chemometrics. Chemom Intell Lab Syst 58:109–130
Roy K, Kar S, Das RN (2015) Statistical methods in QSAR/QSPR in a primer on QSAR/QSPR modeling: fundamental concepts. Springer, Cham
Roy K, Mitra I, Kar S, Ojha PK, Das RN, Kabir H (2012) Comparative studies on some metrics for external validation of QSPR models. J Chem Inf Model 52:396–408
Roy K, Das RN, Ambure P, Aher RB (2016) Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom Intell Lab Syst 152:18–33
Akarachantachote N, Chadcham S, Saithanu K (2014) Cutoff threshold of variable importance in projection for variable selection. Int J Pure Appl Math 94:307–322
Rücker C, Rücker G, Meringer M (2007) Y-randomization and its variants in QSPR/QSAR. J Chem Inf Model 47:2345–2357
Todeschini R, Consonni, V (2009) Molecular descriptors for chemoinformatics: volume I: alphabetical listing/volume II: appendices, references (Vol. 41). John Wiley & Sons
Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O (2016) Applicability domain for QSAR models: where theory meets reality. IJQSPR 1:45–63
Roy K, Ambure P, Kar S (2018) How precise are our quantitative structure–activity relationship derived predictions for new query chemicals? ACS Omega 3:11392–11406
Code availability
The DTC Lab software tools are available at http://teqip.jdvu.ac.in/QSAR_Tools/DTCLab/. Contact kunal.roy@jadavpuruniversity.in for further details.
Funding
PD thanks the Indian Council of Medical Research, New Delhi, for awarding with a Senior Research Fellowship.
Author information
Authors and Affiliations
Contributions
PD: Model development and validation, original draft. KR: Supervision, concept, and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
This is a computational paper not requiring any ethics approval.
Consent to participate
All co-authors have consent to participate in this project.
Consent for publication
All co-authors have consent to publish the results.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
De, P., Roy, K. QSAR and QSAAR modeling of nitroimidazole sulfonamide radiosensitizers: application of small dataset modeling. Struct Chem 32, 631–642 (2021). https://doi.org/10.1007/s11224-021-01734-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-021-01734-w