
Abstract
The [3+2] cycloaddition (32CA) reaction between furoxancarbonitrile oxide (FNO 2) and electron-deficient 2,2,2-trichloroacetonitrile (TCAN 3) in the presence of chloroform was studied within the Molecular Electron Density Theory (MEDT), at the DFT-B3LYP/6-311G(d,p) computational level. This zwitterionic-type (zw-type) 32CA reaction takes place in a highly chemo- and regioselective manner, yielding oxadiazole 4 as the sole product of the reaction, in excellent agreement with the experimental findings. The very low polar character of this zw-type 32CA reaction accounts for the high activation barrier found for this 32CA reaction. A topological analysis of the electron localization function (ELF) over some relevant points of the reaction path permits establishing that this zw-type 32CA reaction takes place along a non-concerted two-stage one-step molecular mechanism. The ELF topological analysis evidences that formation of the C1–N8 and O3–C7 single bonds take place through the sharing of the part of the electron density of the N8 nitrogen and that of the O3 lone pairs toward, respectively, the C1 and C7 pseudoradical centers created along the reaction path.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
[3+2] cycloaddition (32CA) reactions are well known as a powerful and versatile synthetic route to generate five-membered heterocyclic compounds in a high regio- and stereoselective manner. In a 32CA reaction a three-atom-component (TAC), including four π-electrons delocalized over three adjacent nuclei, interacts with an unsaturated bond to yield corresponding [3+2] cycloadduct [1,2,3]. In terms of their geometry, TACs can be classified as allylic-type (A-TACs), with a bent structure such as nitrones (Nis), and propargylic-type (P-TACs), with a linear structure such as nitrile oxides (NOs) [4]. It is worthy to note that 32CA reactions can take place in a desirable regio- and stereoselective fashion when TACs and unsaturated skeletons are electronically activated employing appropriate functional groups [5].
On the basis of theoretical approaches, chemical reactivity of TACs in 32CA reactions can be correlated with the ground state (GS) electronic structure of participating TACs. In this sense, Ess and Houk proposed a distortion/interaction model (DIM) in which distortion of reactants (TAC as well as unsaturated bond) within going from GS electronic structure toward transition state (TS) structure and, then, interaction between distorted reactants at TS is required to generate corresponding [3+2] cycloadduct [6]. In terms of participation, twelve TACs in 32CA reaction toward ethylene and acetylene, Ess and Houk found a good linear correlation coefficient (R2 = 0.97) between B3LYP/6-31G(d) computed activation and distortion energies. It should be noted that, in this model, distortion energy for TAC and/or unsaturated bond is calculated through removing corresponding fragment at TS geometry with no taking into account this very important fact that “the external potential created by one fragment over the other one is lost when one of two interacting fragments is removed” [7]. In other words, dividing the TS geometry into two separate fragments has no physical sense and, thus, DIM not only is not able to rationalize the geometry dependence of activation energy [2, 8] but also seems to need a serious revision. On the other hand, a new reactivity model has been recently introduced by Domingo. In this model, namely Molecular Electron Density Theory study (MEDT), it is emphasized that “while distribution of the electron density is responsible for the molecular shape and physical properties, the capability for changes in electron density, and not the molecular orbital (MO) interactions, is responsible for the reactivity” [9]. MEDT, in addition to exploration reaction paths, takes the analysis of the conceptual density functional theory (CDFT) indices [10], quantum topological analysis of the electron localization function (ELF) [11], quantum theory of atoms in molecules (QTAIM) [12] analysis, and non-covalent interaction (NCI) [13] analysis into consideration to study the molecular reactivity in organic reactions in a rigorous manner [14].
Several MEDT studies have been directed to different 32CA reactions resulting in a reasonable and straightforward classification for this type of cycloadditions as (i) pseudo(di)radical-type (pdr-type); (ii) pseudo(mono)radical-type (pmr-type); (iii) carbenoid-type (cb-type); and (iv) zwitterionic-type (zw-type) 32CA reactions [15]. Such a helpful classification enables us to predict the reactivity of a given TAC in the corresponding 32CA reaction in terms of ELF analysis over the GS electronic structure of participating TAC. In this way, a 32CA reaction of type (i) and (ii) includes TACs with a pseudo(di)radical and pseudo(mono)radical structure, respectively, while in a 32CA reaction of type (iii) and (iv) TACs with a carbenoid and zwitterionic structure are involved, respectively. Scheme 1 shows the Lewis structures derived from ELF analysis over the GS electronic structure of the TACs and corresponding reactivity type in 32CA reactions [14, 15].

Lewis structures derived from ELF analysis over GS electronic structure of the TACs and corresponding reactivity type in 32CA reactions
It is worth mentioning that the reactivity of TACs is decreased from pdr-type toward zw-type (left to right in Scheme 1) 32CA reactions. Indeed, a pdr-type 32CA reaction can easily take place via an earlier TS, even if the reaction does not provide a considerable polarity, while a zw-type 32CA reaction requires proper nucleophilic/electrophilic activation to take place in a polar fashion with a noticeable rate [15].
NOs, as mentioned, belong to the P-TACs whose 32CA reaction toward C–C double bonds yields isoxazolines with noticeable synthetic and biological applications [16]. Isoxazolines are sufficiently stable and can be converted into α,β-unsaturated ketones [17], β-hydroxycarbonyl compounds [18], and 1,3-aminoalcohols [19] through functionalization or ring-cleaving processes. Recently, the reactivity, regioselectivity, and molecular mechanism aspects of 32CA reaction between NOs and C–C double bonds have been theoretically studied by Domingo et. al [20, 21]. The use of a C–N triple bond as the ethylene component in a 32CA reaction toward NOs leads to construction of oxadiazoles possessing a diversity of useful biological effects [22]. The large impact of oxadiazole derivatives on drug discovery across some disease areas such as cancer [23], obesity [24], infection [25], and diabetes [26] has also been well proved.
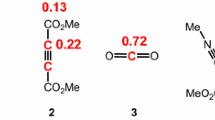
Very recently, the 32CA reaction of furoxancarbonitrile oxide (FNO) toward the C–N triple bond has experimentally been investigated by Larin and co-workers in which nitration of furoxancarbaldehyde oxime 1 followed by thermolysis of the formed nitrolic acid furnishes FNO 2. Then, in situ generated FNO 2 participates in a chemo- and highly regioselective 32CA reaction toward electron-deficient 2,2,2-trichloroacetonitrile, TCAN 3, to produce 1,2,4-oxadiazole 4 in a moderate yield 56% (see Scheme 2) [27].

Synthesis of 1,2,4-oxadiazole 4 over the course of chemo- and regioselective 32CA reaction of in situ generated FNO 2 toward electron-deficient TCAN 3, experimentally investigated by Larin and co-workers [27]
The main goal of the present investigation is to perform an MEDT study over 32CA reaction between FNO 2 and electron-deficient TCAN 3 to shed light on the energetics, chemo- and regioselectivity, and molecular mechanism of this reaction. It is worthy to indicate that, to the best of our knowledge, 32CA reaction of an NO toward C–N triple bond has not ever been studied from molecular mechanism (the bond forming/breaking patterns point of view).
Details of calculations
The B3LYP functional [28] together with the standard 6-311G(d,p) [29] basis set was employed within full geometry optimizations and, then, optimized stationary points were characterized by frequency calculations at the same level of theory in order to ensure all reactants and products have not any imaginary frequency and TSs have one and only one true imaginary frequency along the reaction coordinate. Employing the second order González–Schlegel integration method [30, 31], the intrinsic reaction coordinate (IRC) paths [32] were traced in both forward and backward directions to ensure located TSs truly connect two associated minima. The wavefunction stability of optimized FNO 2 and TSs was also examined using “STABLE” keyword.
Solvent effects of chloroform were implicitly applied through re-optimization of the gas phase located stationary points using polarizable continuum model (PCM) [33, 34] followed by frequency calculation at 339.0 K to obtain thermochemical functions. Note that since the studied 32CA reaction (see Scheme 2) is experimentally refluxed for 2 h in the presence of chloroform, temperature of 339.0 K which is 5° higher than the normal boiling point of chloroform seems to be a reasonable choice to apply thermal effects over thermochemical functions.
Natural atomic charges, calculated through natural population analysis (NPA) [35, 36], were taken into consideration to evaluate the value of global electron density transfer (GEDT) [37] as a measure of the polar character of the studied 32CA reaction. CDFT global reactivity indexes as well as Parr functions [38] were computed using the equations described in reference 9. Employing TopMod software package [39], ELF analyses were carried out over the B3LYP/6-311G(d,p) generated monodeterminantal wavefunctions. All computations were performed by means of Gaussian 09 revision D.01 [40].
Results and discussion
The present MEDT study is given in the four different sections as follows: (1) in “ELF and NPA analysis over GS electronic structure of FNO 2,” the GS electronic structure of FNO 2 is characterized by means of ELF as well as NPA analyses to have a deep insight over the reactivity of FNO 2 in 32CA reactions; (2) in “Analysis of the global and local CDFT reactivity indices at the GS electronic structure of FNO 2 and TCAN 3,” CDFT reactivity indices at the GS electronic structure of FNO 2 and TCAN 3 are analyzed; (3) in “Exploration of the reaction paths involved in the interaction between FNO 2 and TCAN 3,” the competitive reaction paths involved in the interaction between FNO 2 and TCAN 3 are explored to shed light over the energetics of reaction; and (4) finally, in “Elucidation of molecular mechanism in 32CA reaction between FNO 2 and TCAN 3 via the ELF topological analysis,” ELF analysis over the most relevant points along the IRC profile corresponded to the energetically most favorable TS involved in 32CA reaction of FNO 2 toward TCAN 3 enables us to extract bond forming/breaking patterns portraying molecular mechanism aspects in details.
ELF and NPA analysis over GS electronic structure of FNO 2
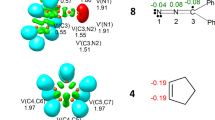
Since FNO 2 includes two different NO and Ni frameworks, any of which can potentially be involved in a zw-type 32CA reaction toward TCAN 3, an ELF topological analysis of the electron density of FNO 2 was performed in order to characterize the electronic structure of these fragments to obtain an obvious portray about their reactivity. ELF valence attractor positions and corresponding populations, natural atomic charges, and proposed Lewis structures for FNO 2 is given in Fig. 1. As can be seen, the C1–N2 bonding region in FNO 2 is characterized with the presence of two V(C1,N2) and V′(C1,N2) disynaptic basins with a total population of 6e portraying a triple C1–N2 bond. While N2 does not display any V(N2) monosynaptic basin associated with its lone pair, N2-O3 bonding region is distinguished with the existence of one V(N2,O3) disynaptic basin integrating 1.71e which evidently defines a single N2–O3 bonding in FNO 2. Moreover, the existence of two V(O3) and V′(O3) monosynaptic basins with a total population of 5.65e implies that there is a non-bonding region around O3 oxygen atom equivalent to three lone electron pairs. A similar analysis for the Ni framework (C4–N5–O6 bonding region) in FNO 2 shows that the presence of one V(C4,N5) disynaptic basin integrating 3.92e defines a double C4–N5 bond as a part of resonance structure in the aromatic five-membered ring.

Representation of ELF valence attractor positions together with corresponding populations (black values in e), natural atomic charges (blue values in e), and proposed Lewis structure for the GS electronic structure of FNO 2
The presence of one V(N5,O6) disynaptic basin with a population of 1.60e indicates a single N5–O6 bonding region, while the presence of two V(O6) and V′(O3) monosynaptic basins with a total population of 5.70e characterizes a non-bonding region around O6 oxygen atom associated with three lone pairs. On the other hand, the absence of any V(N5) monosynaptic basin clearly indicates that the N5 nitrogen has no lone pair, as a consequence of its delocalization on the aromatic ring.
Once the bonding pattern of FNO 2 was established, the charge distribution was analyzed through an NPA. The atomic charges located over nuclei involved in both NO (C1, N2, and O3 atoms) and Ni (C4, N5, O6 atoms) fragments in FNO 2 are given in Fig. 1. While O3 with a charge of − 0.34e and O6 with a charge of − 0.37e are the most negative centers in NO and Ni frameworks, respectively, the N2 and N5 atoms display the most positive charge of 0.22e and 0.36e, respectively. Such evidences indicate both N2–O3 and N5–O6 are polarized toward oxygen atoms and, at the first glance, a commonly accepted 1,2-zwitterionic structure with a noticeable charge separation may be concluded for NO and Ni frameworks in FNO 2, as depicted in Scheme 2. It should be noted that “within the DFT framework, the charge distribution distinguished by the NPA is the consequence of the asymmetric electron density distribution resulting from the presence of different nuclei in the molecule, rather than the consequence of the resonance Lewis structures” [21]. On the other hand, considering FNO 2 as an integrated molecular system, the most negative center is located over carbon atom of the methyl substituent with a considerable negative charge of − 0.60e (see Fig. 1). In consequence, as shown by the Lewis structure in Fig. 1, the 1,2-zwitterionic representation should be avoided for NO and Ni frameworks in FNO 2 [41]. On the basis of ELF patterns, neither pseudo(mono)radical and pseudo(di)radical character nor carbenoid one (see Scheme 1) is found over both NO and Ni frameworks in FNO 2 and, thus, this species can only participate in a zw-type 32CA reaction toward an unsaturated bond, whether the NO or Ni frameworks of this TAC is involved.
Analysis of the global and local CDFT reactivity indices at the GS electronic structure of FNO 2 and TCAN 3
Global reactivity indexes defined within the CDFT [10, 42] are frequently used as a highly useful tool to describe the reactivity in cycloaddition reactions. Taking this fact into account that the global electrophilicity and nucleophilicity values are scaled based on B3LYP/6-31G(d) computations, FNO 2 and TCAN 3 were fully optimized at this computational level. The global reactivity indices for FNO 2 and TCAN 3, i.e., electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω), and global nucleophilicity (N) are collected in Table 1.
The electronic chemical potential μ of FNO 2, − 4.71 eV, is greater than that of TCAN 3, − 5.61 eV, indicating that along a polar 32CA reaction, the GEDT should take place from FNO 2 toward TCAN 3 which act as nucleophile and electrophile, respectively. FNO 2 and TCAN 3 exhibit a high global electrophilicity index of 2.35 and 2.23 eV, respectively, being classified as a strong electrophilic species within the electrophilicity scale [43]. On the other hand, the almost low global nucleophilicity index of 2.05 eV permits FNO 2 to be located on the borderline between weak and moderate nucleophile within the nucleophilicity scale [44]. It is worth mentioning that the presence of a highly electron-withdrawing CCl3 group at TCAN 3 leads to a negative nucleophilicity index, − 0.02 eV, implying TCAN 3 does not provide any nucleophilic character.
In spite of the high electrophilic character of TCAN 3 arising from existence of highly electron-withdrawing CCl3 functional group, the low nucleophilic character of FNO 2 makes this zw-type 32CA reaction to have a low polar character, demanding a high activation barrier to take place (see latter).
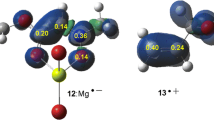
When an electrophile/nucleophile pair come close together, provided steric effects do not interfere, the most electrophilic center of electrophile approaches the most nucleophilic center of nucleophile to proceed the reaction within energetically most preferred channel leading to generation major regioisomer under kinetically controlled conditions. Proposed by Domingo, the electrophilic\( {P}_k^{+} \)and nucleophilic\( {P}_k^{-} \) Parr functions [38] are found as a powerful tool in the study of the local reactivity in polar processes. It is highly important to note that in the definition of Parr functions, the excess of spin electron density reached via the GEDT process from the nucleophile to the electrophile is taken into account as a key factor. Accordingly,\( {P}_k^{-} \) Parr functions of FNO 2 and the electrophilic\( {P}_k^{+} \) Parr functions of TCAN 3 were analyzed in order to characterize the most electrophilic and nucleophilic centers of this species involved in an intramolecular 32CA reaction (see Fig. 2).

3-D representations of the B3LYP/6-311G(d,p) computed Mulliken atomic spin density of the radical cation FNO 2·− (left) and of the radical anion TCAN 3·+ (right), together with the nucleophilic \( {P}_k^{-} \) Parr functions of FNO 2 and the electrophilic \( {P}_k^{+} \) Parr functions of TCAN 3
Analysis of the nucleophilic\( {P}_k^{-} \) Parr functions of FNO 2 indicated that the O6 oxygen belonging to the Ni framework is the most nucleophilic center of this molecules, \( {P}_k^{-} \)= 0.42, while the C1 carbon belonging to the NO framework is a marginally activated nucleophilic center, \( {P}_k^{-} \)= 0.04. On the other hand, analysis of the electrophilic\( {P}_k^{+} \) Parr functions of TCAN 3 indicates that the three chlorines are the most electrophilic centers of this molecules while the C7 carbon is marginally activated as an electrophilic center, \( {P}_k^{+} \)= 0.03. Note that the N8 nitrogen of TCAN 3 possessing a negative \( {P}_k^{+} \) value of − 0.01 is deactivated as an electrophilic center [40].
From the CDFT analysis performed in this section, we can conclude that low nucleophilic charter of FNO 2 together with the poor nucleophilic activation of the NO framework of FNO 2 and the low electrophilic activation of the nitrile framework of TCAN 3 point to a non-polar character and, consequently, a high activation energy for this 32CA reaction.
Exploration of the reaction paths involved in the interaction between FNO 2 and TCAN 3
Upon in situ generation, FNO 2 in the reaction mixture, four competitive 32CA reaction paths toward TCAN 3 can take place as a consequence of the presence of the NO and the Ni frameworks in FNO 2, and the non-symmetry of both reagents. Although the 32CA reaction involving the Ni framework is expected to be very unfavorable because of its participation in the aromatic ring, in excellent agreement with the experimental outcomes [27], the two competitive reaction paths are analyzed to elucidate a quantitative description of the energy profiles and mechanistic aspects. Noted that due to lineal geometry of TCAN 3 no stereoisomeric reaction paths are involved in this 32CA reaction. Scheme 3 displays reaction paths involved in the aforementioned 32CA reactions.

Competitive reaction paths involved in the 32CA reaction of FNO 2 toward TCAN 3. The B3LYP/6-311G(d,p) computed relative enthalpies (red values) and Gibbs free energies (blue values) in the presence of chloroform at 339.0 K and 1.0 atm are given in kcal/mol
As demonstrated in Scheme 3, C1–N2–O3 involvement of FNO 2 toward C–N triple bond of TCAN 3 leads to generation oxadiazole 4 via C1–N8 attack passing through TS1 or generation oxadiazole 5 via C1–C7 attack passing through TS2. On the other hand, cycloadduct 6 can be produced if C4–N5–O6 framework of FNO 2 is involved in 32CA reaction with TCAN 3 via C4–C7 attack which demands to overcome TS3. Similarly, cycloadduct 7 can also be obtained if the C4 carbon in FNO 2 approaches N8 atom of TCAN 3 which requires to overcome TS4. Analysis of the IRC profiles of located TS1, TS2, TS3, and TS4 reveals that formation of all cycloadducts takes place through a one-step mechanism without formation of any stable intermediate.
From relative enthalpies given in Scheme 3, it is obvious that among located TSs, TS1 with an activation barrier of 18.7 kcal/mol is the less energetic TS. Note, however, that the significant activation barrier of TS1 is a consequence of the non-polar character of the zw-type 32CA reaction (see “Analysis of the global and local CDFT reactivity indices at the GS electronic structure of FNO 2 and TCAN 3”), and can be overcome under harsh conditions employed experimentally leading to the formation of oxadiazole 4. On the other hand, TS2 is located by 10.5 kcal/mol over TS1, in clear agreement with the complete regioselectivity observed experimentally. Moreover, the high thermodynamic stabilization gained via the newly formed aromatic five-membered ring in oxadiazole 4 is responsible for the high energy content of 43.0 kcal/mol released within formation of oxadiazole 4 through a quite irreversible pathway. Computed relative enthalpies, however, clearly demonstrate that formation of cycloadducts 6 and 7 which demand to overcome a very high activation barrier of 42.9 (TS3) and 26.2 (TS4) kcal/mol, resulting from the loss of the aromatic character of five-membered ring in FNO 2, should completely be ruled out. These results, in excellent agreement with the experimental findings [25], explain why interaction between FNO 2 and TCAN 3 leads to the formation of oxadiazole 4 as the only isolable product over the course of a chemoselective (C1–N2–O3 involvement rather than C4–N5–O6 one in FNO 2) and regioselective (C1–N8 attack instead of C1–C7 one) 32CA reaction.
The aromatic character of newly formed five-membered rings at oxadiazole 4 and 5 was evaluated using nucleus-independent chemical shift (NICS) method. It has been well documented that the zz component of NICS tensor at 1.0 Å above the ring center, NICS(1)zz, is the most appropriate index to describe the π-orbitals contribution to the aromaticity in an aromatic ring [45]. The B3LYP/6-311++G(d,p) value of NICS(1)zz for the newly formed five-membered ring in cycloadduct 4 and 5 is − 19.5 and − 24.3 ppm, respectively. When these values are compared with that of furan (− 27.2 ppm), as the well-known aromatic five-membered species, it is evidenced that not only is the oxadiazole ring in compounds 4 and 5 highly aromatic but the aromatic character of latter is also significantly greater than the former. Despite such greater aromaticity, cycloadduct 5 is noticeably less stable than 4 by 26.5 kcal/mol (see Scheme 3). In fact, the aromaticity predominance of 5 over 4 is extremely affected by the energy content released within formation of different bonds.
Inclusion of the entropy effects (T∆S) among enthalpy changes leads to a large positive shift within Gibbs free energy values as a consequence of the unfavorable negative entropy associated with these bimolecular processes (see Scheme 3). Despite such large positive shift in the relative Gibbs free energy changes, oxadiazole 4 is the only reachable product under experimentally employed conditions passing through TS1 with a considerable activation Gibbs free energy of 33.0 kcal/mol within a highly exergonic pathway.
The B3LYP/6-311G(d,p) optimized structure of TS1 through TS4 involved in 32CA reactions of FNO 2 toward TCAN 3 including some key geometrical distances as well as the unique imaginary frequency, in cm−1, in the presence of chloroform is given in Fig. 3. Considering that the C–O, C–N, and N–O single-bond formation takes place at different distances, these TSs correspond to asynchronous single-bond formation processes in which the formation of the C–O or C–N single bond involving the C1 carbon atom of TCAN 3 is more advanced than the other single bond (see latter).

B3LYP/6-311G(d,p) optimized structure of TSs involved in 32CA reactions of FNO 2 toward TCAN 3 in the presence of chloroform. While some geometrical distances are given in Angstrom, the unique imaginary frequency given in the parenthesis is in cm−1
Numerous MEDT studies have shown a very good correlation between the polar character and the feasibility of cycloaddition reactions. Accordingly, the polar nature of this 32CA reaction was evaluated by computing the GEDT at the corresponding TSs. Reactions with the GEDT values of 0.00e correspond to non-polar processes, while values higher than 0.20e correspond to polar processes. The B3LYP/6-311G(d,p) GEDT value, which fluxes from the NO framework to the TCAN one is 0.08e at TS1 and 0.11e at TS2. Moreover, the GEDT value which fluxes from the Ni framework to the TCAN one is 0.09e at TS3 and 0.08e at TS4. The low GEDT value found at the energetically most preferred TS1 indicates that this zw-type 32CA reaction has a very low polar character.
Elucidation of molecular mechanism in 32CA reaction between FNO 2 and TCAN 3 via the ELF topological analysis
In order to understand molecular mechanism involved in 32CA reaction between FNO 2 and TCAN 3, ELF topological analysis of some relevant points along the IRC profile of the energetically most favorable TS1 connecting separate FNO 2 and TCAN 3 with oxadiazole 4 was performed.
Figure 4 represents the B3LYP/6-311G(d,p) IRC profile of energetically most preferred TS1 involved in 32CA reaction between FNO 2 and TCAN 3 to generate cycloadduct 4 which includes a total 177 points with a narrow step size of 0.02 Bohr. Position of the most relevant points P1 through P5 is also given along this IRC profile. As depicted in Fig. 4, five relevant points are characterized along the IRC profile of TS1 any of which comprises distinguished change(s) in the ELF valence attractor positions and their populations enabling us to portray bond forming/breaking patterns within interaction between FNO 2 and TCAN 3. Figure 5 displays the ELF valence attractor positions and their populations for separate FNO 2 and TCAN 3 (the top box) and for points P1 through P5. While a detailed explanation about the ELF valence attractors of isolated FNO 2 is given in Fig. 1, the C7–N8 bonding region in the isolated TCAN 3 is characterized with the presence of three V(C7,N8), V′(C7,N8), and V′′(C7,N8) disynaptic basins presenting a total population of 4.53e (see the top box in Fig. 5), which is considerably lower from the expected value of 6e associated with a triple bond.

B3LYP/6-311G(d,p) IRC profile of energetically most preferred TS1 involved in 32CA reaction between FNO 2 and TCAN 3 to generate cycloadduct 4 involving position of the most relevant points P1 through P5

ELF valence attractor positions and their populations for the isolated FNO 2 and TCAN 3 (top box) and, for the most relevant points, P1 through P5 involved in the C1–N8 and C7–O3 single bonds formation along the IRC profile of TS1
This behavior can be related to the polarization of C7–N8 bonding region toward C7 carbon atom due to presence of highly electron-withdrawing CCl3 group, in one hand, and the more electronegative character of the N8 nitrogen than the C7 carbon, in the other hand. Such polarization produces that the population of the C7–C9 single bond becomes 2.28e, and that of the N8 lone pair becomes 3.15e. At point P1, d(C1–N8) = 2.243 Å and d(O3–C7) = 2.297 Å, the first relevant ELF topological changes are evidenced (see Fig. 5). Indeed, the formation of a new V(C1) monosynaptic basin over the C1 carbon, integrating 0.48e, and a new V(N2) monosynaptic basin over the N2 nitrogen, integrating 1.94e, are observed. Consequently, at P1, the C1 pseudoradical center required for the subsequent C1–N8 single-bond formation is already created, while a N2 lone pair has emerged because of depopulation C1–N2 bonding region toward C1 and N2 centers. At P2, d(C1–N8) = 1.939 and d(O3–C7) = 2.037 Å, the electron population of C7–N8 bonding region decreases from the initial value of 4.53e in the separate TCAN 3 to 4.0e.
Such a decrease results in the formation of a new V(C7) monosynaptic basin integrating 0.24e. Consequently, at P2, the C7 pseudoradical center required for the subsequent O3–C7 single-bond formation is already created. At this point, the population of the V(N8) monosynaptic basin has been increased by 0.35e from isolated TCAN 3. At P3, d(C1–N8) = 1.753 Å and d(O3–C7) = 1.882 Å, the presence of a V(C1,N8) disynaptic basin integrating 0.97e indicates that the formation of the first C1–N8 single bond has already begun at a C–N distance of 1.73 Å through the sharing of the electron density of the V(C1) monosynaptic basin and some electron density of the V(N8) monosynaptic basin present at P2. Note that the population of N8 lone pair is significantly decreased from 3.40e at P2 to 2.98e at P3 (see Fig. 5). At P4, d(C1–N8) = 1.619 Å and d(O3–C7) = 1.754 Å, the non-bonding electron density of the O3 oxygen present at P3 is shared into three monosynaptic basins, V(O3), V′(O3), and V′′(O3). The V′′(O3) monosynaptic basins, which integrate 0.43e, will be involved in the formation of the second O3–C7 single bond. Finally, at P5, d(C1–N8) = 1.532 Å and d(O3–C7) = 1.643 Å, the presence of a new V(C7,O3) disynaptic basin with an initial population of 1.11e indicates that the formation of second O3–C7 single bond has already been formed at a O–C distance of 1.64 Å through the sharing of the electron density of the V(C7) monosynaptic basin and that of the V″(O3) monosynaptic basin present at P4.
It is highly important to mention that when the very delayed formation of the O3–C7 single bond starts at point P5 at the short O–C distance of 1.64 Å, the electron population of the already formed C1–N8 single bond reaches 1.62e. This value which is more than 85% of its population at oxadiazole 4 (1.90e) permits establishing that this 32CA takes place through a non-concerted two-stage one-step mechanism [46] in which formation of the second O3–C7 single bond begins when the formation of the first C1–N8 bond becomes almost complete. Moreover, in terms of ELF analysis, the bonding changes along 32CA reaction of FNO 2 toward TCAN 3 are not concerted but sequential, which permits to reject the proposed pericyclic mechanism in which a concerted movement of electrons around a cycle is suggested [43]. Note that the non-concerted two-stage one-step character, portrayed for the molecular mechanism of the studied 32CA reaction, is in quite agreement with previous studies devoted to 32CA reactions [1, 3, 5, 8, 47,48,49,50].
Conclusions
The chemo- and regioselective 32CA reaction of in situ generated FNO 2 with TCAN 3 yielding 1,2,4-oxadiazole 4, experimentally reported very recently by Larin and co-workers [27], has been theoretically studied within the MEDT at the DFT-B3LYP/6-311G(d,p) computational level. The present MEDT study permits to point out some conclusions as follows:
-
Among the four types of reactivity provided for 32CA reactions, the 32CA reaction of FNO 2 toward TCAN 3 should be classified as a zw-type 32CA reaction, in which the low nucleophilic character of FNO 2 is responsible for the non-polar character displayed by the reaction. This non-polar character of this accounts for the high activation enthalpy computed for the reaction, 18.7 kcal/mol.
-
Exploration of the four competitive reaction paths evidently indicates that the NO framework of FNO 2 rather than its Ni one participates in this zw-type 32CA reaction toward TCAN 3. Indeed, while a 32CA reaction at NO framework allows a new aromatic five-membered ring to be generated in oxadiazole 4, the participation of Ni framework requires to loss of the aromaticity in FNO 2 and, thus, it is highly prevented. Consequently, the 32CA reaction of FNO 2 toward TCAN 3 yields oxadiazole 4 as the sole product through a high activation barrier but a very exergonic pathway acting as a driving force, in excellent agreement with the experimental findings.
-
In quite agreement with the previous mechanistic considerations devoted to 32CA reactions, the ELF analysis of the most relevant points located over the IRC profile of the energetically most favorable TS1 characterizes a non-concerted two-stage one-step molecular mechanism for the studied 32CA reaction.
-
In this sense, while formation of the first C1–N8 single bond takes place through the sharing of part of the electron density of the N8 lone pair with that of the C1 pseudoradical center, formation of the second O3–C7 single bond takes place through the sharing of part of the electron density of the O3 lone part with that of the C7 pseudoradical center. Both C1 and C7 pseudoradical centers are created in this zw-type 32CA reaction along the reaction path through the depopulation of the C1–N2 and C7–N8 bonding regions of the reagents.
-
The bonding changes along this 32CA reaction evidenced by the ELF analysis make it possible to rule out the proposed pericyclic mechanism in which a concerted movement of electrons around a cycle is suggested.
References
Emamian S (2010) Generation of a substituted 1,2,4-thiadiazole ring via the [3+2] cycloaddition reaction of benzonitrile sulfide toward trichloroacetonitrile. A DFT study of the regioselectivity and of the molecular mechanism. C R Chim 18:1277–1283
Emamian S, Lu T, Moeinpour F (2015) Can the high reactivity of azomethine betaines in [3 + 2] cycloaddsition reactions be explained using singlet-diradical character descriptors?. What molecular mechanism is actually involved in these cycloadditions? RSC Adv 5:62248–62259
Emamian S (2017) A molecular electron density theory study of [3+2] cycloaddition reaction between azomethine ylides and electron-deficient nitroalkenes. Chemistry Select 2:4193–4203
Gothelf KV, Jorgensen KA (1998) Asymmetric 1,3-dipolar cycloaddition reactions. Chem Rev 98:863–910
Emamian S (2015) Understanding the regioselectivity and molecular mechanism in the synthesis of isoxazoles. Understanding the regioselectivity and molecular mechanism in the synthesis of isoxazoles containing pentafluorosulfanyl substitution via a [3+2] cycloaddition reaction: a DFT study. J Fluor Chem 178:165–172
Ess DH, Houk KN (2008) Theory of 1,3-dipolar cycloadditions: distortion/interaction and frontier molecular orbital models. J Am Chem Soc 130:10187–10198
Domingo LR, Ríos-Gutiérrez M, Duque-Noreña M, Chamorro E, Pérez P (2016) Understanding the carbenoid-type reactivity of nitrile ylides in [3+2] cycloaddition reactions towards electron-deficient ethylenes: a molecular electron density theory study. Theor Chem Accounts 135:160–172
Emamian S (2016) How the mechanism of a [3 + 2] cycloaddition reaction involving a stabilized N-lithiated azomethine ylide toward a π-deficient alkene is changed to stepwise by solvent polarity? What is the origin of its regio- and endo stereospecificity? A DFT study using NBO, QTAIM, and NCI analyses. RSC Adv 6:75299–75314
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319–1324
Domingo LR, Ríos-Gutérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748–770
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5043
Bader RWF (1990) Atoms in molecules: a quantum theory. Claredon Press, Oxford, U.K
Johnson ER, Keinan Mori-Sanchez SP, Contreras-Garćıa J, Cohen J, Yang AW (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506
Domingo LR, Ríos-Gutérrez M, Pérez P (2017) How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A molecular electron density theory study. Tetrahedron 73:1718–1724
Domingo LR, Ríos-Gutérrez M (2017) A molecular electron density theory study of the reactivity of azomethine imine in [3+2] cycloaddition reactions. Molecules 22:750–770
Kozikowski AP (1984) The isoxazoline route to the molecules of nature. Acc Chem Res 17:410–416
Jäger V, Grund H (1976) Eliminative ring opening of 2-Isoxazolines: a new route to α,β-unsaturated ketones. Angew Chem Int Ed 15:50–51
Curran DP (1978) Reduction of .DELTA.2-isoxazolines: a conceptually different approach to the formation of aldol adducts. J Am Chem Soc 104(1982):4024–4026
Jäger V, Buss V , Schwab W, Syntheses via isoxazolines III Diastereoselective synthesis of γ-amino-alcohols with 2 and 3 chiral centres. Tetrahedron Lett 19:3133–31360
Domingo LR, Emamian S, Salami M, Ríos-Gutiérrez M (2016) Understanding the molecular mechanism of [3+2] cycloaddition reaction of benzonitrile oxide toward an N-vinylpyrrole derivative with the aid of ELF topological analysis. J Phys Org Chem 29:368–376
Ndassa IM, Adjieufack AAI, Mbadcam Ketcha J, Berski S, Ríos-Gutiérrez M, Domingo LR (2017) Understanding the reactivity and regioselectivity of [3+2] cycloaddition reactions between substituted nitrile oxides and methyl acrylate. A molecular electron density theory study. Int J Quantum Chem https://doi.org/10.1002/qua.25451
Kadi AA, El-Brollosy NR, Al-Deeb OA, Habib EE, Ibrahim TM, El-Emam AA (2007) Synthesis, antimicrobial, and anti-inflammatory activities of novel 2-(1-adamantyl)-5-substituted-1,3,4-oxadiazoles and 2-(1-adamantylamino)-5-substituted-1,3,4-thiadiazoles. Eur J Med Chem 42:235–242
Zhang H-Z, Kasibhatla S, Kuemmerle Kemnitzer JW, Ollis- Mason K, Qiu L, Crogan-Grundy C, Tseng B, Drewe J, Cai SX (2005) Discovery and structure−activity relationship of 3-aryl-5-aryl-1,2,4-oxadiazoles as a new series of apoptosis inducers and potential anticancer agents. J Med Chem 48:5215–4223
Lee SH, Seo HJ, Lee SH, Jung ME, Park JH, Park HJ, Yoo J, Yun H, Na J, Kang SY, Song KS, Kim MA (2008) Biarylpyrazolyl oxadiazole as potent, selective, orally bioavailable cannabinoid-1 receptor antagonists for the treatment of obesity. J Med Chem 51:7216–7233
Cottrell DM, Capers J, Salem MM, DeLuca-Fradley K, Croft SL, Werbovetz KA (2004) Antikinetoplastid activity of 3-aryl-5-thiocyanatomethyl-1, 2, 4-oxadiazoles. Bioorg Med Chem 12:2815–2824
Boström J, Hogner A, Llinàs A, Wellner E, Plowright AT (2012) Oxadiazoles in medicinal chemistry. J Med Chem 55:817–1830
Larin AA, Fershtat LL, Ananyev IV, Makhova NN (2017) Versatile approach to heteroarylfuroxan derivatives from oximinofuroxans via a one-pot, nitration/thermolysis/[3+ 2]-cycloaddition cascade. Tetrahedron Lett 42:3993–3997
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
González C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527
González C, Schlegel HB (1991) Improved algorithms for reaction path following: higher-order implicit algorithms. J Chem Phys 95:5853–5856
Fukui F (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094
Simkin BY, Sheikhet I (1995) Quantum chemical and statistical theory of solutions-a computational approach. Ellis Horwod, London
Reed AE, Weinstock RB, Weinhold FF (1985) Natural population analysis. J Chem Phys 83:735–746
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494
Noury S, Krokidis K, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–605
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani BV, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JF, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09 (Revision D.01). Gaussian, Inc., Wallingford CT
Adjieufack AI, Ndassa IM, Ketcha Mbadcam J, Ríos-Gutiérrez M, Domingo LR (2017) Steric interactions controlling the syn diastereofacial selectivity in the [3+2] cycloaddition reaction between acetonitrile axide and 7-oxanorborn-5-en-2-ones. A molecular electron density theory study. J Phys Org Chem 30:e3710
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874
Domingo LR, Aurell M, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common dienedienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4442
Jarmillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct (THEOCHEM) 865:68–72
Fallah-Bagher-Shaidaei H, Wannere CS, Corminboeuf C, Puchta R, Schleyer PVR (2006) Which NICS aromaticity index for planar π rings is best? Org Lett 8:863–866
Domingo LR, Saéz JA, Zaragozá RJ, Arnó M (2008) Understanding the participation of quadricyclane as nucleophile in polar 2 sigma+2 sigma+2 pi cycloadditions toward electrophilic pi molecules. J Organomet Chem 73:8791–8799
Ríos-Gutiérrez M, Chafaa F, Nacereddine AK, Djerourou A, Domingo LR (2016) A DFT study of [3+2] cycloaddition reactions of an azomethine imine with N-vinyl pyrrole and N-vinyl tetrahydroindole. J Mol Graph Model 70:296–304
Ríos-Gutiérrez M, Darù A, Tejero T, Domingo LR, Merino P (2017) A molecular electron density theory study of the [3 + 2] cycloaddition reaction of nitrones with ketenes. Org Biomol Chem 15:1618–1627
Domingo LR, Pérez P, Ortega DE (2013) Why do five-membered heterocyclic compounds sometimes not participate in polar Diels-Alder reactions? J Organomet Chem 78:2462–2471
Carey FA, Sundberg RJ (2000) Advanced organic chemistry. Part A: structure and mechanisms. Springer, New York
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Hosseini, S.J., Emamian, S. & Domingo, L.R. Participation of furoxancarbonitrile oxide in [3+2] cycloaddition reaction toward C–N triple bond: a Molecular Electron Density Theory study of regioselectivity and mechanistic aspect. Struct Chem 30, 317–326 (2019). https://doi.org/10.1007/s11224-018-1203-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1203-4