
Abstract
A molecular electron density theory (MEDT) study is performed for the [3 + 2] cycloaddition (32CA) reactions of benzonitrile oxide (BNO) and diphenyldiazomethane (DPDM) to cyclopentene (CP) and norbornene (NBN) with the objective to analyse the experimentally observed acceleration in NBN reactions relative to the CP ones. The activation enthalpy of the 32CA reaction of NBN with BNO is lowered than that of CP by 2.1–2.9 kcal mol−1 in gas phase, DMSO, acetonitrile and THF, while the corresponding differences are 1.3–1.8 kcal mol−1 with DPDM. The 32CA reactions of DPDM show lower activation parameters compared to that of BNO consistent with the respective pseudo(mono)radical and zwitterionic type characters of DPDM and BNO. The syn diastereofacial approach of NBN is energetically feasible compared to the anti one. The global electron density transfer (GEDT) is identified to anticipate the minimal electron density flux at the TS entity, which is otherwise not accounted by the global electrophilicities of the separated regents. This MEDT study allows comprehending that for these non-polar reactions, the acceleration in NBN cycloadditions compared to the CP ones is due to the relatively lower energy cost demanded for the depopulation and subsequent rupture of C–C double bond of NBN followed by sequential bonding changes along the reaction paths, rather than the “predistorted geometry towards the TSs” as previously proposed in the 32CA reactions of norbornene derivatives.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Norbornene (NBN) (bicyclo [2.2.1] hep-2-ene) and its derivatives have established their promising practical applications in polymer science, solar energy converters, medicinal and agricultural chemistry over more than 60 years [1]. Very recently, the copolymerisation of NBN and divinyl benzene has been reported for the synthesis of graft polymers [2], while NBN derivatives have also found several current applications in bioorthogonal reactions [3, 4]. The unique angularly strained structural geometry of NBN fosters its synthetic applicability as an attractive candidate in organic synthesis [1, 5,6,7,8,9,10]. The behaviour of NBN derivatives in ring opening metathesis polymerisation [5, 6], cycloaddition reactions [7], Wagner–Meerwein rearrangement [8], Prins reaction [9] and photochemical excitation [10] are well documented for synthetic applications.
In 2015, Truong et al.[7] reported the first application of the [3 + 2] cycloaddition (32CA) reaction of nitrile oxide 1 with NBN 2 to prepare hydrogels (Scheme 1). In 2016, Zhang et al. [11] performed the polyaddition of azide-containing NBN-based monomer for polymer resin industries. Several other applications [1] have placed the 32CA reactions of NBN derivatives in the top-shelf priority of applied chemistry and consequently invited chemists to explore the possibilities, design new strategies and analyse these reactions.

32CA reaction of nitrile oxide 1 with NBN 2 as a cross-linking step in the preparation of hydrogels
In the late 1950s, the 32CA reactions of diazoalkanes and aryl azides with NBN derivatives attracted attention for the first time [12] while in 1965, the 32CA reactions of aryl azides with NBN 2 were reported [13]. The proposed mechanism involving 1,5-zwitterions [12] was thereafter questioned to keep pace with these experimental outcomes, subsequently proposing the concerted mechanism without intermediates [12, 14].
Huisgen [14] compared the reactivities of cyclopentene CP 4 and NBN 2 towards benzonitrile oxide BNO 5, respectively, leading to the products 6 and 7 (Scheme 2) and towards diphenyl diazomethane DPDM 8 leading to the products 9 and 10, respectively (Scheme 2). In both cases, the 32CA reaction of NBN 2 showed faster reaction compared to that of CP 4 [14].

32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
Theoretically, the 32CA reactions of azides and norbornene NBN 2 have been analysed in terms of distortion–interaction theory, in which the activation energy is partitioned into “distortion” and “interaction” terms [15]. Recently, we have proposed the theoretical alternative to distortion–interaction theory for the analysis of strain-promoted azide–alkyne cycloaddition (SPAAC) reactions [16] based on the molecular electron density theory (MEDT) perspective proposed by Domingo in 2016 [17,18,19]. The decisive role of electron density changes for easy depopulation of the alkyne triple bond region along the SPAAC reactions rather than the distorted transition geometry model was thus identified [16]. MEDT studies for 32CA reactions of acetonitrile oxide and 7-oxanorbornen-5-en-2ones were reported in 2017 [20], and very recently, we have applied MEDT to explain the unexpected reactivity of electrophilic diazoalkanes towards norbornadiene [21]. In addition to strain promotion, the selectivities and catalytic effects can be successfully comprehended within the MEDT framework [22,23,24,25,26,27,28].
Although norbornadiene selectivity has been addressed using the MEDT concept, the experimentally observed acceleration in norbornene NBN 2 cycloadditions relative to cyclopentene has not been accounted so far, while the distortion–interaction theory has been applied to study the azide–norbornene cycloadditions [15]. Herein, we present the MEDT study for the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2 (Scheme 2) experimentally realised by Huisgen [14] to analyse the observed acceleration in NBN cycloadditions. The use of MPWB1K functional with the 6-311G(d,p) basis set has been recently recommended as a useful and precise computational model for the analysis of 32CA reactions [22] justifying its applicability for the present study. (1) In Sect. 2.1, the electron localisation function [29, 30] (ELF) of the reagents was studied to classify the three atom components (TACs) and correlate the electronic structure with molecular reactivity [22]. (2) In Sect. 2.2, the conceptual density functional theory [31,32,33,34] (CDFT) reactivity descriptors were analysed to predict the polar character. (3) In Sect. 2.3, the potential energy surface (PES) along the feasible reaction paths was followed to obtain the energy profile (4) In Sect. 2.4, the ELF study of the intrinsic reaction coordinate [35] (IRC) points along the energetically favoured reaction paths is analysed to identify the catastrophe [36] and thereafter structure the plausible mechanism (5) In Sect. 2.5, the ELF at the TSs is analysed (6) In Sect. 2.6, the intermolecular interaction at the TSs is analysed by quantum theory of atoms-in-molecules (QTAIM [37,38,39]) parameters.
2 Computational methods
The reactants, TSs and products were optimised using MPWB1K functional [40] in conjunction with the 6-311G(d,p) basis set [41]. The Berny’s analytical gradient optimisation method [42] was applied for the optimisation of reactants, transition states and products. Frequency calculations were performed to characterise and verify the optimised stationary points as minima or TSs. The minimum energy reaction path connecting the optimised minima through the located TSs was verified by intrinsic reaction coordinate [35] (IRC) calculations using Gonzales–Schlegel integration method [43, 44].
The conceptual density functional theory [31, 32] (CDFT) indices, electronic chemical potential [45] (μ), global hardness [46] (η), electrophilicity [33] (ω) and nucleophilicity [34] (N) indices were calculated using Eqs. (1–4).
where EHOMO and ELUMO are the HOMO and LUMO energies; and EHOMO(tetracyanoethylene) considered as the reference for nucleophilicity index [34] represents the HOMO energy of tetracyanoethylene. Note that the B3LYP/6-31G(d) level was used for CDFT calculations since the standard electrophilicity and nucleophilicity scales are defined at this computational level [33, 34].
The global electron density transfer [47] (GEDT) at the TSs was calculated using Eq. (5). The charges were calculated by natural population analysis [48, 49].
The asymmetry index [50] ∆l at the interacting centres A and B of the forming bonds in the TSs was calculated using Eq. (6)
where rTSA−B and rPA−B are the distances between A and B in the TSs and the products, respectively.
Solvent effects in DMSO, acetonitrile and THF were studied using polarisable continuum model [51, 52] (PCM) modelled within the self-consistent reaction field [53,54,55] (SCRF). All reactants, transition states and products were optimised in the respective solvents at PCM/MPWB1K/6-311G(d,p) level of theory and the minima and TSs were characterised by frequency calculations. The thermodynamic parameters were calculated at 1 atm pressure and 298 K in gas phase, DMSO, acetonitrile and THF.
All optimisations, frequency calculations and IRC studies were performed using Gaussian 16 suite of programs [56].
The ELF [29, 30] and QTAIM [37,38,39] parameters were calculated using Multiwfn software [57] with high-quality grid. The ELF localisation domains were visualised using UCSF Chimera software [58].
2.1 ELF Topological analysis of the reactants BNO 5, DPDM 8, CP 4 and NBN 2
In several MEDT studies [17,18,19,20,21,22,23,24,25,26,27,28], the ELF topological study has allowed a reasonably good correlation of the electronic structures with the molecular reactivity consistent with the standard classification of the TACs into pseudodiradical [59], pseudo(mono)radical [23], carbenoid [60] and zwitterionic type [22]. Accordingly, the ELF at the ground-state electronic structures of BNO 5 and DPDM 8 is studied to correlate the electronic structure and the reactivity of the reagents. The ELF localisation domains, basin attractor positions and the most significant valence basin populations are given in Fig. 1.

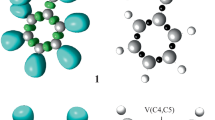
MPWB1K/6-311G(d,p) ELF basin attractor positions of BNO 5, DPDM 8, CP 4 and NBN 2 and the proposed Lewis-like structures together with the natural atomic charges in average number of electrons e. Negative, negligible and positive charges are shown in red, green and blue colours, respectively
The ELF topological analysis of BNO 5 shows the presence of four monosynaptic basins V(O1), V′(O1), V″(O1) and V′′′(O1) integrating a total population of 5.68 e associated with the non-bonding electron density at O1 oxygen; two disynaptic basins V(C3,N2) and V'(C3,N2) integrating 5.92 e associated with the C3-N2 triple bond; one disynaptic basin V(N2,O1) integrating 1.72 e associated with the N2–O1 single bond. The absence of pseudoradical or carbenoid centres in BNO 5 allows its classification as a zwitterionic TAC participating in zw-type 32CA reactions associated with high activation energy barrier [17,18,19, 22].
The ELF topological analysis of DPDM 8 shows the presence of two monosynaptic basins V(C3) and V′(C3) integrating 1.02 e, associated with a pseudoradical centre at C3; two disynaptic basins V(C3,N2) and V′(C3,N2) integrating 3.06 e associated with the C3-N2 double bond; two disynaptic basins V(N1,N2) and V′(N1,N2) integrating 3.72 e associated with the N1–N2 double bond; two monosynaptic basins V(N1) and V′(N1) integrating 3.82 e associated with the non-bonding electron density on N1 nitrogen. The presence of pseudoradical centre at C3 classifies DPDM 8 as a pseudo(mono)radical TAC [23], participating in pmr-type 32CA reactions.
ELF topology of CP 4 and NBN 2 shows the presence of a pair of disynaptic basins V(C4,C5) and V′(C4,C5) integrating a total population of 3.48 e and 3.45 e, associated with the C4–C5 double bond.
The proposed Lewis-like structures and the natural atomic charges are given in Fig. 1. Oxygen O1 is negatively charged by − 0.42 e while N2 and C3 show almost similar positive charges 0.22 e and 0.20 e in BNO 5. The pseudoradical carbon C3 and the N1 and N2 nitrogen nuclei show negligible charges in DPDM 8, which is different from the commonly accepted 1,2-zwitterionic concept of the diazoalkanes. The C4 and C5 carbon of CP 4 and NBN 2 are negatively charged by 0.19 e and 0.20 e, respectively.
2.2 Analysis of the CDFT indices of reactants BNO 5, DPDM 8, CP 4 and NBN 2
The use of global reactivity descriptors defined within the CDFT [31,32,33,34] is a well-established tool to predict the reactivity of reagents participating in 32CA reactions. The B3LYP/6-31G(d) calculated global reactivity indices, namely the electronic chemical potential μ, chemical hardness η, electrophilicity ω and nucleophilicity N indices of the reagents, are given in Table 1.
The electronic chemical potential μ of BNO 5 (μ = − 3.81 eV) and DPDM 8 (μ = − 3.35 eV) is lower than that of CP 4 (μ = − 2.71 eV) and NBN 2 (μ = − 2.79 eV), suggesting that along a polar process, the electron density will flux from CP 4 and NBN 2 to BNO 5 and DPDM 8 via reverse electron density flux (REDF) reactions [61].
The chemical hardness η of BNO 5 (η = 5.01 eV) and DPDM 8 (η = 3.70 eV) are lower than that of CP 4 (η = 7.21 eV) and NBN 2 (η = 6.94 eV). Thus, the TACs, BNO 5 and DPDM 8 are softer and more prone to electron density deformation compared to CP 4 and NBN 2.
Within the electrophilicity scale [33], BNO 5 (ω = 1.45 eV) is classified as a moderate electrophile, while DPDM 8 (ω = 1.52 eV) as a strong electrophile. On the other hand, CP 4 (ω = 0.51 eV) and NBN 2 (ω = 0.56 eV) are classified as marginal electrophiles with ω < 0.80 eV.
The analysis of CDFT indices predicts that along a polar process, BNO 5 and DPDM 8 will behave as the electrophilic species, while CP 4 and NBN 2 as the nucleophilic ones. The global hardness of BNO 5 and DPDM 8 show lower values, indicating more resistance to exchange electron density compared to CP 4 and NBN 2. Note that CP 4 and NBN 2 show higher electronic chemical potentials predicting higher propensity to exchange electron density compared to BNO 5 and DPDM 8.
Although CP 4 and NBN 2 show marginal electrophilicity, they are classified as the moderate nucleophiles on the nucleophilicity scale similar to BNO 5, while DPDM 8 is classified as a strong nucleophile. Note that although B3LYP/6-31G(d) and MPWB1K/6-311G(d,p) reactivity indices (given in Supplementary Material) show different values, similar trends of reactivity are predicted.
2.3 Analysis of the energy profile of the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
Due to the molecular symmetry of CP 4, only one reaction path is feasible for the 32CA reactions with BNO 5 and DPDM 8. These two reactions follow one step mechanism, allowing location of the TSs TS1 and TS4 leading to isoxazoline 6 and pyrazoline 9, respectively (Scheme 3).

32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
Two competitive stereoisomeric reaction paths are feasible for the 32CA reactions of NBN 2 with BNO 5 and DPDM 8. They are related to the syn and anti approach modes of BNO 5 and DPDM 8 along the two stereoisomeric faces of the C–C double bond of NBN 2. For the 32CA reaction of BNO 5 with NBN 2, the syn and anti reaction paths lead to two diastereomeric isoxazolines 7 and 11, respectively, via TS2 and TS3 while for the 32CA reaction of DPDM 8 with NBN 2, the syn and anti approaches lead to two diastereomeric isoxazolines 10 and 12, respectively, via TS5 and TS6 (Scheme 3). The relative electronic energies, enthalpies, entropies and Gibbs free energies of the TSs and the cycloadducts are given in Table 2.
A series of appealing conclusions can be proposed from the energy profile. (1) The 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2 show the reaction Gibbs free energies between − 24.0 (9) and − 40.0 (7) kcal mol−1 in gas phase, − 25.7 (9) and − 40.5 (7) kcal mol−1 in DMSO and acetonitrile and − 25.3 (9) and − 40.4 (7) kcal mol−1 in THF, suggesting highly exergonic reactions which makes them irreversible. (2) The activation energy of TS4 is lowered than that of TS1 by 3.6 kcal mol−1 in gas phase and 4.1 kcal mol−1 in DMSO, acetonitrile and THF. The higher activation energy of the zw-type reaction of BNO 5 than that of the pmr-type reaction of DPDM 8 is the expected molecular reactivity in agreement with the TAC characterisation [17,18,19, 21, 23]. (3) For the 32CA reaction of BNO 5 with NBN 2, the syn approach mode is energetically favoured than the anti one with the activation energy of TS2 lowered than that of TS3 by 6.0 kcal mol−1 in gas phase, 6.2 kcal mol−1 in DMSO and acetonitrile and 6.1 kcal mol−1 in THF. Similarly, the 32CA reaction of DPDM 8 with NBN 2 also prefers the syn approach mode, than the anti one with the activation energy of TS5 lowered than that of TS6 by 5.6 kcal mol−1 in gas phase and 5.7 kcal mol−1 in DMSO, acetonitrile and THF. The free energy profiles in gas phase are shown in Fig. 2. Inclusion of solvent effects shows minimal impact on the energy results in each case.

Relative free energies (kcal mol−1) of gas-phase-optimised reactants, TSs and products along the syn and anti reaction paths for the 32CA reactions of NBN 2 with BNO 5 and DPDM 8
(4) The activation energy of TS2 is lowered than that of TS1 by 2.1 kcal mol−1 in gas phase, 2.4 kcal mol−1 in DMSO and acetonitrile and 2.3 kcal mol−1 in THF. The activation energy of TS4 is lowered than that of TS5 by 1.4 kcal mol−1 in gas phase and 1.6 kcal mol−1 in DMSO, acetonitrile and THF. These differences account for the experimentally observed [14] faster 32CA reactions of BNO 5 and DPDM 8 with NBN 2 compared to the analogous reactions with CP 4. The free energy profile in gas phase is shown in Fig. 3.

Relative free energies (kcal mol−1) of gas-phase-optimised reactants, TSs and products for the 32CA reactions of CP 4 and NBN 2 (syn path) with BNO 5 and DPDM 8
(5) Thermodynamic corrections to the activation energies at 298 K increase the activation enthalpies between 0.3 and 1.9 kcal mol−1, while the reaction enthalpies are decreased between 3.1 and 4.8 kcal mol−1. Inclusion of entropies to the enthalpies rises the activation free energies between 12.1 and 13.6 kcal mol−1 in gas phase, 12.7 and 15.2 kcal mol−1 in DMSO and acetonitrile and 11.4 and 14.9 kcal mol−1 in THF, while the reaction free energies are decreased between 13.2 and 15.4 kcal mol−1 in gas phase, 11.6 and 14.9 kcal mol−1 in DMSO, 12.0 and 15.2 kcal mol−1 in acetonitrile and 12.5 and 15.3 kcal mol−1 in THF. The gas-phase activation free energies for 32CA reaction of BNO 5 with CP 4 and NBN 2 are 29.0 and 27.4 kcal mol−1, while that for the 32CA reaction of DPDM 8 with CP 4 and NBN 2 is 25.9 and 24.6, respectively (Fig. 3).
The gas-phase-optimised geometries of TS1–TS6 are given in Fig. 4, while the bond lengths and asymmetry indices ∆l are listed in Table 3. (1) For the 32CA reactions of BNO 5, the C3–C4 forming bond length is longer than that of the forming O1–C5 bond in TS1, TS2 and TS3. However, the respective asymmetry index [50] ∆l values 0.122, 0.142 and 0.157 suggest earlier C–C bond formation than the O–C one in each case (Table 3). The bond order predicted earlier C–C bond formation complies with the BET analysis (Sect. 2.4). For the 32CA reactions of DPDM 8, the C–C bond formation is slightly earlier than the N–C one with ∆l values of 0.038 and 0.011, respectively, for TS4 and TS6, suggesting a hardly asynchronous process. Inclusion of solvent effects does not considerably modify the gas-phase geometries.

MPWB1K/6-311G(d,p) optimised geometries (gas phase) of TS1–TS6 associated with the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
Now considering that the C–C bond formation begins at 2.0–1.9 Å [18] and the O–C/N–C bond formation at 1.9–1.8 Å [18], it is evident that the C–C, O–C and N–C covalent bond formation has not started at the TSs showing bond distances greater than 2.0 Å, in agreement with the ELF study (Sect. 2.5) and QTAIM analysis (Sect. 2.6) at the TSs.
In 1999 [62], Domingo performed DFT studies for Diels–Alder (DA) reactions of nitroethylenes and observed decrease in the activation energies with the nucleophilicity of the ethylene derivatives. Subsequently, the influence of polarity on the reaction feasibility was quantitatively established in 2003 from the correlation of the activation parameters with global electrophilicity ω index for the DA reactions of cyclopentadiene and cyanoethylenes [63]. A reasonable good correlation (R2 = 0.99) between the charge transfer (CT) at the TSs and the experimental rate constants was finally reported by Domingo in 2009 [64], revealing the predominant role of electron density flux at the TSs, termed as the global electron density transfer [47] (GEDT) in 2014. Polar reactions are characterised by GEDT values above 0.20 e, while non-polar reactions show values less than 0.10 e. Higher GEDT values are indicative of more polarity and hence faster reactions. In 2017, Domingo [65] reported that the higher GEDT values lead to depopulation of the C–C double bond and easy rupture of C–C bonds in cycloaddition reactions resulting in the lower activation parameters of polar CAs relative to the non-polar ones.
The calculated GEDT at the TSs are given in Fig. 4. The GEDT values are found between 0.02 and 0.05 e, suggesting non-polar reactions and are classified as null electron density flux [61] (NEDF). Interestingly, the minimal electron density flux predicted by GEDT is contrary to the CDFT predicted strong electrophilicities of BNO 5 and DPDM 2 and marginal electrophilicities of CP 4 and NBN 2 (Table 1). Similar prediction has also been observed recently for the 32CA reactions of substituted diazoalkanes (DAAs) to norbornadiene [21], where the 32CA reaction is experimentally decelerated due to the introduction of electron-withdrawing substituents in the simplest diazoalkane, although the DAAs were predicted as the electrophilic counterpart to norbornadiene by CDFT indices, while the GEDT predictions agree well with the experiments. Thus, for the 32CA reactions of BNO 5 and DPDM 8 to CP 4 and NBN 2, it seems that the electron density fluxes minimally between the reacting counterparts at the TS entity and this local phenomenon is not anticipated in the global reactivity indices defined within the CDFT calculated for the separated reagents. On the other hand, GEDT predicts this local electron density flux at the TS entity, classifying the reactions as the non-polar ones. From the BET study (Sect. 2.4), it is further evident that the electronic flux between reacting counterparts differ minimally for NBN and CP reactions at each nuclear configuration along the reaction path, and the experimentally observed acceleration in NBN ones is the outcome of the relatively lower energy cost (EC) demanded for the C–C double bond rupture, as suggested by Domingo for non-polar CAs [21, 65], thus complying with the GEDT prediction. Note that the calculated high activation energies (Table 2) also characterise these non-polar reactions and the QTAIM analysis (Sect. 2.5) shows non-covalent interactions at the TSs.
2.4 BET study for the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
Bonding evolution theory (BET) proposed by Krokoidis [66] allows structuring the plausible mechanism of chemical reactions [67]. Within the MEDT framework, BET allows to study the electron density changes along the reaction path. Accordingly, the BETs for the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2 were studied. For the 32CA reactions of DPDM 8, the energetically preferred syn approach mode was selected for the study. Detailed BET studies are given in Sections S1–S4 of Supplementary Material.
The 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2 can be differentiated into eight ELF topological phases with the representative IRC points S0–S7 characterising the beginning of each phase. The IRC points are denoted with numerals I and II to designate 32CA reactions of BNO 5 with CP 4 and NBN 2, respectively, while III and IV numerals indicate the 32CA reactions of DPDM 8 with CP 4 and NBN 2, respectively. The simple representations of the molecular mechanisms for the 32CA reactions of CP 4 with BNO 5 and DPDM 8 are given in Schemes 4 and 5.

Simple representation of the molecular mechanism of the 32CA reaction of BNO 5 with CP 4

Simple representation of the molecular mechanism of the 32CA reaction of DPDM 8 with CP 4
The ELF of the starting points S0-I, S0-II, S0-III and S0-IV of Phase I is similar to that of the separated reagents (Fig. 1).
Phase II of the 32CA reactions is identified with the formation of monosynaptic basin V(N2) associated with the non-bonding electron density at N2 nitrogen. Note that the energy cost (EC) to reach phase II are 11.5 and 10.4 kcal mol−1 for the 32CA reaction of BNO 5 with CP 4 and NBN 2, respectively, while the corresponding ECs are 8.5 and 8.8 kcal mol−1, respectively, for the 32CA reactions of DPDM 8.
Phase III of the 32CA reactions of BNO 5 with CP 4 and NBN 2 is associated with the formation of pseudoradical centre at C3 carbon, which demands EC of 13.8 and 12.2 kcal mol−1, respectively. Owing to the pseudo(mono)radical character of DPDM 8, the pseudoradical centre at C3 carbon already exists in the separated reagent, which consequently demands no extra EC to form along the reaction path.
Phase IV for the 32CA reactions of BNO 5 with CP 4 and NBN 2 is associated with the rupture of the C4–C5 double bond demanding EC of 15.2 and 13.3 kcal mol−1, respectively, while the similar bonding change is observed in phase III for the 32CA reactions of DPDM 8 demanding EC of 11.0 and 9.8 kcal mol−1 for CP 2 and NBN 2. TS1 and TS2 belong to phase IV, while TS4 and TS5 belong to phase III of the respective 32CA reaction. Thus, the activation energies for 32CA reactions of BNO 5 are associated with the formation of non-bonding electron density at N2 nitrogen, pseudoradical centre at C3 and rupture of the C4–C5 double bond, while those of DPDM 8 are associated with the formation of non-bonding electron density of N2 and rupture of the C4–C5 double bond. This explains the lower EC calculated for the 32CA reactions of DPDM 8 compared to that of BNO 5. Note that the EC demanded for the rupture of C–C double bond in the 32CA reactions of BNO 5 and DPDM 8 with CP 4 are higher than that with NBN 2 by 1.9 and 1.2 kcal mol−1, respectively, leading to accelerated NBN reactions, as observed experimentally.
In the later phases along the reaction path, the rupture of C4–C5 double bonds of CP 4 and NBN 2 is followed by the formation of pseudoradical centres at C4 and C5 carbons and the coupling of the pseudoradical centres at C3 and C4 carbons to form the first C3–C4 single bond in all four reactions. Subsequently, the pseudoradical centre at C5 couples with the part of non-bonding electron density of O1 oxygen of BNO 5 and N1 nitrogen of DPDM 8 to form the second O1–C5 and N1–C5 covalent bonds in the respective reactions.
Note that the earlier C3–C4 bond formation is predicted in complete agreement with the asymmetry index calculations at the TSs (Table 3). For 32CA reactions of BNO 5 with CP 4 and NBN 2, the formation of the second covalent O1–C5 bond begins when the first C3–C4 bond has been 86% and 88% completed, suggesting a highly asynchronous process of bond formation. For 32CA reactions of DPDM 8 with CP 4 and NBN 2, the formation of the second covalent N1–C5 bond begins when the first C3–C4 bond has been 75% and 81% completed, suggesting a comparatively less asynchronous bond formation. The minimal electronic flux between the reacting counterparts along the reaction paths suggests null electron density flux [61] (NEDF), classifying the non-polar character of these 32CA reactions.
2.5 ELF topological analysis at the TSs associated with the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
ELF topological study at the TSs associated with the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2 was performed to comprehend the electronic structure. The ELF valence basin populations at TS1–TS6 are given in Table 4, while the ELF localisation domains and basin attractor positions of TS1, TS2, TS4 and TS5 are represented in Fig. 5.

ELF localisation domains (isovalue = 0.84) and basin attractor positions of the TS1, TS2, TS4 and TS5
TS1, TS2 and TS3 associated with the 32CA reactions of BNO 5 show monosynaptic basins V(O1), V′(O1) and V″″(O1) (in TS2) integrating total population of 5.62–5.63 e associated with the non-bonding electron density at O1 oxygen. TS4, TS5 and TS6 associated with the 32CA reactions of DPDM 8 show monosynaptic basin V(N1) integrating total population of 3.63–3.65 e associated with the non-bonding electron density at N1 nitrogen. TS1–TS6 show the presence of monosynaptic basin V(N2) integrating 1.92–2.05 e associated with the non-bonding electron density at N2 nitrogen, which is not present in BNO 5 and DPDM 8.
TS1, TS2 and TS3 show the presence of monosynaptic basin V(C3) integrating 0.81 e, 0.79 e and 0.37 e, respectively, associated with the pseudoradical centre at C3 carbon, which is not present in BNO 5. On the other hand, the two monosynaptic basins V(C3) and V'(C3) present in DPDM 8 have merged into one monosynaptic basin V(C3) integrating 0.83 e, 0.84 e and 0.82 e, respectively, in TS4, TS5 and TS6 associated with the pseudoradical centre at C3.
TS1–TS6 show the presence of V(C3,N2) disynaptic basin associated with the C3–N2 bonding region. Note that the C3–N2 bonding region is depopulated from 5.92 e in BNO 5 to 1.56 e in TS1 and TS2, and 1.94 e in TS3 to create pseudoradical centre at C3 carbon and non-bonding electron density at N2 nitrogen. The C3–N2 bonding region in DPDM 8 is depopulated from 3.06 to 1.96 e in TS4 and 1.98 e in TS5 and TS6 to create non-bonding electron density at N2 nitrogen.
TS1–TS6 show the presence of disynaptic basin V(C4,C5) integrating 3.18–3.23 e associated with the C4–C5 bonding region. Note that the two disynaptic basins V(C4,C5) and V′(C4,C5) integrating 3.48 e and 3.45 e in CP 4 and NBN 2 have merged into one disynaptic basin V(C4,C5) indicating rupture of the C4–C5 double bond at the TSs.
Finally, the TSs do not show the presence of disynaptic basins associated with the formation of new single bonds suggesting that the formation of C3–C4 or O1/N1–C5 covalent bonds has not been started, consistent with the forming bond distances greater than 2 Å (Table 3).
2.6 Analysis of interatomic interactions at the TSs associated with the 32CA reactions of BNO 5 and DPDM 8 with CP 4 and NBN 2
The nature of interatomic interactions can be quantitatively assessed from the calculation of QTAIM parameters [37,38,39]. For the present study, CP1 and CP2 indicate the bond critical points (BCPs) of the forming C3–C4 and O1/N1–C5 bonds in the TSs. The total electron density ρ and the Laplacian of electron density \(\nabla ^{2} \rho (r_{c} )\) at CP1 and CP2 in the TSs are given in Table 5.
The calculated total electron density ρ is less than 0.1 a.u. in each case (0.052–0.058 e at CP1 and 0.020–0.065 e at CP2) suggesting rather non-covalent interactions evident from the positive Laplacian of electron density \(\nabla ^{2} \rho (r_{c} )\) (0.044–0.054 e at CP1 and 0.087–0.099 e at CP2). The formation of new C3–C4 and O1/N1–C5 covalent bonds has not been started at the TSs, consistent with the ELF study at the TSs discussed (Sect. 2.5).
3 Conclusion
This MEDT report analyses the experimentally observed acceleration observed in the 32CA reactions of norbornene NBN 2 to benzonitrile oxide BNO 5 and diphenyldiazomethane DPDM 8 relative to that of cyclopentene CP 4.
The 32CA reactions were highly exergonic and hence irreversible. The pseudo(mono)radical type 32CA reactions of DPDM 8 show lower activation energies compared to that observed in the zwitterionic type 32CA reactions of BNO 5, owing to the presence of pseudoradical centre at C3 of DPDM 8.
The 32CA reactions of NBN 2 with BNO 5 and DPDM 8 along the syn diastereofacial mode are energetically favoured by 6.0 and 5.6 kcal mol−1 in gas phase compared to the anti one. Similar reactivity trend is also observed in DMSO, acetonitrile and THF.
The activation energies for the 32CA reactions of NBN 2 with BNO 5 and DPDM 8 are lowered by 2.1 kcal mol−1 and 1.4 kcal mol−1 in gas phase compared to that of CP 4 complying with the experimentally observed acceleration in NBN cycloadditions. Interestingly, the minimal GEDT at the TSs predicts non-polar character, contrary to the strong electrophilic character of the TACs BNO 5 and DPDM 8 and the marginal electrophilicity of CP 4 and NBN 2. The non-polar character is also evident from the BET study showing minimal electronic flux along the reaction path, while the experimentally observed acceleration of NBN reactions is due to the lower EC demanded for the rupture of the C–C double bond of NBN compared to that of CP. It therefore seems that the local electronic flux at the TS entity is predicted by GEDT, but cannot be accounted by the global electrophilicities and electronic chemical potentials of the separated reagents.
References
Flid VR, Gringolts ML, Shamsiev RS, Finkelshtein ES (2018) Norbornene, norbornadiene and their derivatives: promising semi-products for organic synthesis and production of polymeric materials. Russ Chem Rev 87:1169–1205. https://doi.org/10.1070/RCR4834
Chowdhury S, Tanaka R, Nakayama Y, Shiono T (2020) Synthesis of norbornene/divinylbenzene copolymers catalyzed by anilinonaphthoquinone-ligated nickel complexes and their applications for the synthesis of graft polymers. J Polym Sci 58:1564–1570. https://doi.org/10.1002/pol.20200133
Madl CM, Heilshorn SC (2018) Bioorthogonal strategies for engineering extracellular matrices. Adv Func Mater 28:1706046. https://doi.org/10.1002/adfm.201706046
Zheng M, Zheng L, Zheng P, Li J, Zhang Y (2015) Development of bioorthogonal reactions and their applications in bioconjugation. Molecules 20:3190–3205. https://doi.org/10.3390/molecules20023190
Ivin KJ, Mol JC (1997) Olefin metathesis and metathesis polymerization, 2nd edn. Academic Press, London
Hobbs CE, Lin B, Malinski T (2015) Norbornene derivatives from a metal-free, strain-promoted cycloaddition reaction: new building blocks for ring-opening metathesis polymerization reactions. J Polym Sci Part A Polym Chem 53:2357–2362. https://doi.org/10.1002/pola.27691
Truog VX, Zhou K, Simon GP, Forsythe JS (2015) Nitrile oxide-norbornene cycloaddition as a bioorthogonal crosslinking reaction for the preparation of hydrogels. Marcromol Rapid Commun 36:1729–1734. https://doi.org/10.1002/marc.201500314
Daştan A, Balci M (2005) High temperature bromination. part 18: Bromination of benzonorbornadiene derivatives: polybrominated benzonorbornenes and benzonorbornadienes. Tetrahedron 61:5481–5488. https://doi.org/10.1016/j.tet.2005.03.132
Sauers RR, Sonnet PE (1964) Prins reaction of norbornene. J Org Chem 29:754–755. https://doi.org/10.1021/jo01026a503
Chow YL, Cheng XE (1993) The reaction pattern of norbornene with excited state carbonyl compounds: photochemical preparations of norbornene derivatives. Res Chem Intermed 19:211–234. https://doi.org/10.1163/156856793X00082
Zhang X, Zhang O, Wu Y, Feng C, Xie C, Fan X, Li P (2016) Polyaddition of azide-containing norbornene-based monomer through strain-promoted 1,3-dipolar cycloaddition reaction. Macromol Rapid Commun 37:1311–1317. https://doi.org/10.1002/marc.201600233
Breugst M, Reissig H-U (2020) The Huisgen reaction: milestones of the 1,3-dipolar cycloaddition. Angew Chem Int Ed 59:12293–12307. https://doi.org/10.1002/anie.202003115
Scheiner P, Schomaker JH, Deming S, Libbey WJ, Nowack GP (1965) The addition of aryl azides to norbornene. a kinetic investigation. J Am Chem Soc 87:306–311. https://doi.org/10.1021/ja01080a030
Huisgen R (1963) Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew Chem Int Ed 2:633–645 https://doi.org/10.1002/anie.196306331
Lopez SA, Houk KN (2013) Alkene distortion energies and torsional effects control reactivities, and stereoselectivities of azide cycloadditions to norbornene and substituted norbornenes. J Org Chem 78:1778–1783. https://doi.org/10.1021/jo301267b
Domingo LR, Acharjee N (2020) Unravelling the strain-promoted [3+2] cycloaddition reactions of phenyl azide with cycloalkynes from the molecular electron density theory perspective. New J Chem 44:13633–13643. https://doi.org/10.1039/D0NJ02711A
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319. https://doi.org/10.3390/molecules21101319
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur J Org Chem 2:267–282. https://doi.org/10.1002/ejoc.201800916
Domingo LR, Acharjee N (2020) Molecular Electron Density Theory: A New Theoretical Outlook on Organic Chemistry. In Ul-Haq Z, Wilson AK (ed) Frontiers in Computational Chemistry. Bentham and Science, Singapore 5:174–227 https://doi.org/10.2174/9789811457791120050007
Adjieufack AI, Ndassa IM, Mbadcam JK, Ríos-Gutiérrez M, Domingo LR (2017) Steric interactions controlling the syn stereofacial selectivity in the [3 + 2] cycloaddition reaction between acetonitrile oxide and 7-oxanorborn-5-en-2-ones: a molecular electron density theory study. J Phys Org Chem 30:e3710. https://doi.org/10.1002/poc.3710
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2021) Unveiling the unexpected reactivity of electrophilic diazoalkanes in [3+2] cycloaddition reactions within molecular electron density theory. Chemistry 3:74–93. https://doi.org/10.3390/chemistry3010006
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C,N-Dialkyl Nitrones with ethylene derivatives. J Org Chem 83:2182-2197 https://doi.org/10.1021/acs.joc.7b03093
Domingo LR, Acharjee N (2020) Unveiling the high reactivity of strained dibenzocyclooctyne in [3+2] cycloaddition reactions with diazoalkanes through the molecular electron density theory. J Phys Org Chem 33:e4100. https://doi.org/10.1002/poc.4100
Salim HAM, Acharjee N, Domingo LR, Abdallah HH (2021) A molecular electron density theory study for [3+2] cycloaddition reactions of 1-pyrroline-1-oxide with disubstituted acetylenes leading to bicyclic 4-isoxazolines. Int J Quant Chem 121:e26503. https://doi.org/10.1002/qua.26503
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2019) A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 24:832. https://doi.org/10.3390/molecules24050832
Domingo LR, Acharjee N (2020) A molecular electron density theory study of the Grignard reagent-mediated regioselective direct synthesis of 1,5-disubstituted-1,2,3-triazoles. J Phys Org Chem 33:e4062. https://doi.org/10.1002/poc.4062
Salim HAM, Acharjee N, Abdallah HH (2021) Insights into the mechanism and regioselectivity of the [3 + 2] cycloaddition reactions of cyclic nitrone to nitrile functions with a molecular electron density theory perspective. Theor Chem Acc 140:1. https://doi.org/10.1007/s00214-020-02703-y
Domingo LR, Acharjee N, Salim HAM (2020) Understanding the reactivity of trimethylsilyldiazoalkanes participating in [3+2] cycloaddition reactions towards diethylfumarate with a molecular electron density theory perspective. Organics 1:3–18. https://doi.org/10.3390/org1010002
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403. https://doi.org/10.1063/1.458517
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686. https://www.nature.com/articles/371683a0
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874. https://doi.org/10.1021/cr990029p
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748 https://doi.org/10.3390/molecules21060748
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 58:4417-4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct THEOCHEM 865:68–72. https://doi.org/10.1016/j.theochem.2008.06.022
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163. https://doi.org/10.1021/j100717a029
Thom R (1972) Stabilité Structurelle et Morphogenèse (Interéditions), Paris
Bader RFW (1990) Atoms in molecules: a quantum theory. Clarendon Press, Oxford
Bader RFW, Essén H (1984) The characterization of atomic interactions. J Chem Phys 80:1943–1960. https://doi.org/10.1063/1.446956
Kumar PSV, Raghavendra V, Subramanian V (2016) Bader’s theory of atoms in molecules (AIM) and its applications to chemical bonding. J Chem Sci 128:1527–1536. https://doi.org/10.1007/s12039-016-1172-3
Zhao Y, Truhlar DG (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der waals interactions. J Phys Chem A 108:6908–6918. https://doi.org/10.1021/jp048147q
Hehre WJ, Radom L, Schleyer PVR, Pople J (1986) Ab initio molecular orbital theory. Wiley, New York, USA
Schlegel HB (1982) Optimisation of equilibrium geometries and transition structures. J Comput Chem 3:214–218. https://doi.org/10.1002/jcc.540030212
González C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
González C, Schlegel HB (1991) Improved algorithms for reaction path following: higher-order implicit algorithms. Chem Phys 95:5853–5860. https://doi.org/10.1063/1.461606
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, Oxford
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428. https://doi.org/10.1039/C4RA04280H
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746. https://doi.org/10.1063/1.449486
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926. https://doi.org/10.1021/cr00088a005
Jasiński R (2015) A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett 56:532–535. https://doi.org/10.1016/j.tetlet.2014.12.007
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094. https://doi.org/10.1021/cr00031a013
Simkin BY, Sheikhet I (1995) Quantum chemical and statistical theory of solutions-a computational approach. Ellis Horwood, London
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041. https://doi.org/10.1063/1.474659
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255:327–335. https://doi.org/10.1016/0009-2614(96)00349-1
Barone V, Cossi M, Tomasi J (1998) Geometry optimisation of molecular structures in solution by the polarizable continuum model. J Comput Chem 19:404–417. https://doi.org/10.1002/(SICI)1096-987X(199803)19:4%3c404::AID-JCC3%3e3.0.CO;2-W
Frisch M, Trucks G, Schlegel H, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Petersson G, Nakatsuji H, Gaussian 16. Revision A (2016), 3.
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comp Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng CC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Domingo LR, Chamorro E, Pérez P (2010) Understanding the high reactivity of the azomethine ylides in [3 + 2] cycloaddition reactions. Lett Org Chem 7:432–439. https://doi.org/10.2174/157017810791824900
Ríos-Gutiérrez M, Domingo LR (2019) The carbenoid-type reactivity of simplest nitrile imine from a molecular electron density theory perspective. Tetrahedron 75:1961–1967. https://doi.org/10.1016/j.tet.2019.02.014
Domingo LR, Ríos-Gutiérrez M, Pérez P (2020) A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv 10:15394–15405. https://doi.org/10.1039/D0RA01548B
Domingo LR, Arnó M, Andrés J (1999) Influence of reactant polarity on the course of the inverse-electron-demand Diels-Alder reaction. A DFT study of regio-and stereoselectivity, presence of lewis acid catalyst, and inclusion of solvent effects in the reaction between nitroethene and substituted ethenes. J Org Chem 64:5867–5875. https://doi.org/10.1021/jo990331y
Domingo LR, Aurell MJ, Pérez P, Contreras R (2003) Origin of the synchronicity on the transition structures of polar Diels-Alder reactions. Are these reactions [4 + 2] processes? J Org Chem 68:3884–3890. https://doi.org/10.1021/jo020714n
Domingo LR, Sáez JA (2009) Understanding the mechanism of polar Diels-Alder reactions. Org Biomol Chem 7:3576–3583. https://doi.org/10.1039/B909611F
Domingo LR, Ríos-Gutiérrez M, Pérez P (2017) How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A molecular electron density theory study. Tetrahedron 73:1718–1724. https://doi.org/10.1016/j.tet.2017.02.012
Krokidis X, Noury S, Silvi B (1997) Characterization of elementary chemical processes by catastrophe theory. J Phys Chem A 101:7277–7282. https://doi.org/10.1021/jp9711508
Andrés J, González-Navarrete P, Safont VS, Silvi B (2017) Curly arrows, electron flow, and reaction mechanisms from the perspective of the bonding evolution theory. Phys Chem Chem Phys 19:29031–29046. https://doi.org/10.1039/C7CP06108K
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Acharjee, N., Mohammad-Salim, H.A. & Chakraborty, M. Unveiling [3 + 2] cycloaddition reactions of benzonitrile oxide and diphenyl diazomethane to cyclopentene and norbornene: a molecular electron density theory perspective. Theor Chem Acc 140, 113 (2021). https://doi.org/10.1007/s00214-021-02811-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-021-02811-3