
Abstract
DFT/BP86 calculations have been carried out on a series of hypothetical binuclear compounds of general formula (L3M)2(C12N2H8) (M = Sc–Ni, L3 = (CO)3, (PH3)3 and Cp−, and C12N2H8 = phenazine ligand-denoted Phn). The various structures with syn and anti configurations have been investigated, in order to determine the phenazine’s coordination to first-row transition metals of various spin states with syn and anti conformations. The lowest energy structures depend on the nature of the metal, the spin state, and the molecular symmetry. This study has shown that the electronic communication between the metal centers depends on their oxidation state and the attached ligands. The tricarbonyl and the triphosphine ligands gave rise to comparable results in terms of stability order of isomers, metal-metal bond distances, and the coordination modes. Metal-metal multiple bonding has been evidenced for Sc, Ti, and V complexes to compensate the electronic deficiency. The Cr, Mn, Fe, Co, and Ni-rich metals prefer the anti conformation due to the enhancement of the metal valence electron count. The spin density values calculated for the triplet and quintet spin structures point out that the unpaired electrons are localized generally on the metal centers. The Wiberg bond indices are used to evaluate the metal-metal bonding. Furthermore, calculations using the BP86-D functional which take into account the attractive part of the van der Waals type interaction potential between atoms and molecules that are not directly connected to each other gave comparable results to those obtained by BP86 functional in terms of coordination modes, HOMO-LUMO gaps, metal-metal bond orders, and the stability order between isomers, but with slight deviation of M–C, M–N, and M–M bond distances not exceeding 3%.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The organometallic transition metal complexes of polyarenes grow in importance, where the diversity of their structures arises principally from the flexibility of the PAH (polycylic or heteropolycyclic aromatic hydrocarbons) ligands like phenazine, a molecule providing various coordination sites. The phenazine molecule with 14π electrons with three fused six-membered rings is a bisannulated derivative of pyrazine as a planar aromatic N-heterocyclic ligand and has been widely studied in chemistry and biology [1,2,3,4,5,6]. Furthermore, the variation of the ancillary ligands like as carbonyls, cyclopentadienyl, or phosphines connected to the metals could conduct to significant modifications of the geometrical parameters, chemical, and physical properties of these complexes.
Lately, homobimetallic complexes have attracted great attention in which two equivalent metals are bonded through a hydrocarbon bridge, i.e., bimetallic complexes, and most of the information on the phenomena of electronic communication stemmes from investigation on this category of compounds, whose outcomes have been widely reconsidered henceforth [7,8,9,10]. However, binuclear complexes of the phenazine ligand are less investigated, where few examples of mono- and polymetallic are known experimentally [11,12,13,14,15,16] and are scarcely studied theoretically [17, 18].
In this work, we were interested in the determination of the electronic structure, the coordination mode, and the metal-metal bonding of binuclear [M(L3)2(Phn)] (M = Sc−Ni, L3 = Cp−, (CO)3, (PH3)3 and Phn = phenazine) depending on the metal nature, the spin state, and the auxiliary ligands. In order to get a deeper insight into intermetallic communications in binuclear complexes, we have examined the mutual influence of the metal centers in binuclear complexes with a bridging phenazine. For the all studied complexes, two stable conformations syn and anti, which obey to the metal nature and of the auxiliary ligand attached to the metals, which are presented in Scheme 1.

Configurations (a) and (b) encountered in (L3M2)(Phn) (L3 = Cp and (CO)3) complexes
The importance for transition metal complexes arises from the broad range of geometries which can be adopted owing to the flexibility of the polycyclic aromatic hydrocarbons forming organometallic structures.
Besides, the presence of a large number of transition metals, the variation of the auxiliary ligands such as tricabonyls, cyclopentadienyl, or phosphines bound to the metals or the oxidation state of the metals could provoke substantial variation of the chemical and physical properties in the system and would act differently due to the different binding capabilities of the isolobal (CO)3, (PH3)3, and Cp− ligands arising from differences in their frontier orbitals differing in energy as well as in shape and spatial extent. The interactions between ML3 fragments in binuclear complexes are well discussed in our previous work [17].
The used BP86 method has proven to be valuable to determine the molecular and electronic structures and relative stabilities and reproduce nicely the experimental data for related systems [19,20,21,22,23,24,25,26,27]. For reasons of comparison, the contribution of intramolecular London dispersion effects [28, 29], which take into account the attractive part of the van der Waals type interaction potential between atoms and molecules that are not directly connected to each other have been applied using the BP86-D functional [30].
Computational details
Density functional theory (DFT) calculations were carried out on the studied compounds using the Amsterdam Density Functional (ADF) program version 2014.01 [31], developed by Baerends and co-workers [32,33,34,35,36]. Electron correlation was treated within the local density approximation (LDA) in the Vosko-Wilk-Nusair parametrization [37]. The BP86 functional that combines Becke’s 1988 exchange functional (B) [38, 39] with Perdew’s 1986 gradient corrected correlation functional (P86) [40, 41]. In order to compensate for the incapacity of BP86 functional to describe dispersion effects correctly, the DFT-D method consisting of BP86-D [30] was used for all calculations. The numerical integration procedure applied for the calculations was developed by te Velde et al. [36]. The atom electronic configurations were described by a triple-ζ Slater-type orbital (STO) basis set for H 1 s, C 2 s and 2p, N 2 s, and 2p augmented with a 3d single-ζ polarization for C and N atoms and with a 2p single-ζ polarization for H atoms. A triple-ζ STO basis set was used for the first-row transition metals 3d and 4 s, for Pd 4d and 5 s augmented with a single-ζ 4p polarization function for the first row. A frozen-core approximation was used to treat the core shells up to 1 s for C, N, and 3p for the first-row transition metals [32,33,34,35,36]. Full geometry optimizations were carried out using the analytical gradient method implemented by Versluis and Ziegler [42]. Spin-unrestricted calculations were performed for all the open-shell systems. Frequency calculations [43, 44] were performed on all the studied compounds to check that the optimized structures are at local minima. Representation of the molecular orbitals and molecular structures was done using ADF-GUI [31] and MOLEKEL4.1 [45], respectively. The NAO-based Wiberg bond indices (WBI) [46] are obtained from NBO calculations implemented in NBO 6.0 program using all electron basis sets [47].
Results and discussion
Scandium model complexes
The scandium metal as d3 is the poorest element of the first row of the transition metals. The full geometry optimizations of [(CO)3Sc]2(Phn) species show that each (CO)3Sc metal fragment is bound to the phenazine ligand through an η6-coordiantion mode (Fig. 1) independently of the ancillary ligand and of the spin state. Indeed, the optimized high-quintet states are obtained high in energy than the singlet and the triplet ones whatever the considered configuration; thus they are not discussed in this section. For example, the syn-[(CO)3Sc]2(Phn) quintet structure of conformation (a) is computed less stable by 8.6 and 11.2 kcal/mol than the triplet and singlet structures, respectively. In reference to the Sc−C bond distances in the ranges 2.330–2.662, 2.307–2.725, and 2.518–2.555 Å for singlet and triplet syn conformation (a) and triplet anti conformation (b) structure (Table 1), respectively, are considered as short ones, conducting to the phenazine ligand to be connected to the bimetallic unit either by the two adjacent cycles or by both separated ones. Whatever the adopted configuration, each Sc metal is considered as neutral center obtained for singlet structures, but monocationic for the triplet ones. The syn-[(CO)3Sc]2(Phn) singlet C s structure exhibits small HOMO-LUMO gap of 0.55 eV, for which a double bond is attributed to the Sc−Sc contact, based on the Sc−Sc bond distances of 3.231 against 3.220 Å (BP86-D) and the molecular orbital localizations (Fig. 2) besides the Wiberg indice [46] of 0.39 obtained by NBO program [47, 48]. The 10π-electrons of the coordinated rings are shared equitably by both neutral Sc centers, thus acquiring the 16-MVE configurations.

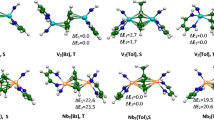
Optimized [(CO)3M]2(ƞ6, ƞ6-Phz) (M = Sc, Ti, V) singlet and triplet structures in their syn and anti configurations. Relative energies ∆E between isomers are given in (kcal/mol). S, T, and Q indicate the singlet, triplet, and quintet spin states

MO diagrams of syn-[(CO)3Sc]2(ƞ6, ƞ6-Phz), syn-[(CO)3Ti]2(ƞ6, ƞ6-Phz), and syn-[(CO)3V]2 (ƞ6, ƞ6-Phz) complexes of singlet state C s symmetry
The [(PH3)3Sc]2(Phn) species possessing 28 MVE (metal valence electrons) like as [(CO)3Sc]2(Phn) ones should adopt the same behavior in terms of geometry, electronic structure, the metal-ligand, and the metal-metal bonding. Indeed, the lowest energy for [(PH3)3Sc]2(Phn) structures corresponding to the syn one of the configuration (a) (Fig. S1) of the Supplementary information, exhibiting η6,η6-coordination mode is computed more stable than other isomers as shown in Table S1 of the supplementary information. Whereas, the Sc–C bond lengths of [(PH3)3Sc]2(Phn) species are shorter than those of [(CO)3Sc]2(Phn) species Table S1. Furthermore, this syn-[(PH3)3Sc]2(Phn) singlet structure of configuration (a) is obtained more stable than those of open-shell triplet and quintet ones by 4.6 and 12.4 kcal/mol, respectively, which display the same η6,η6-coordination mode, but with elongated Sc–Sc, Sc–C, and Sc–N bond distances (Table S1).
However, the [(Cp)Sc]2(Phn) complexes having two less electrons than [(CO)3Sc]2(Phn) and [(PH3)3Sc]2(Phn) should correspond to deficient structures due to the depopulation of π metal-metal MO, in which the metals behave like cationic Sc(I) centers corresponding to 16- and 14-MVE configurations. The lowest energy structure for the [(Cp)Sc]2(Phn) species is a syn triplet state bonded to each (Cp)Sc unit through an η6-coordiantion mode lying lower in energy by 2.2 kcal/mol than the singlet structure (Fig. 2 and Table 2). The syn and anti structures of configuration (b) are found higher in energy than the global minimum by at least 10.5 kcal/mol, exhibiting η6-coordiantion mode of each external C6 ring. The MO diagram sketched in Fig. 4 shows a small HOMO-LUMO gap of 0.52 eV for the [(Cp)Sc]2(Phn) species consistent with the partially occupation of the bonding combinations of the so-called “t 2g” orbitals of the CpM moieties composed of the occupied σ (37a’) and the vacant π (24a”) and δ (38a’) components. These different MO occupations and the Sc-Sc bond distance of 3.456 suggest the presence of a single bond and cationic Sc(I) centers of 14-MVE configuration (Fig. 4). However, the syn and anti-[(Cp)Sc]2(Phn) quintet structures lie at least 22.4 and 35.7 kcal/mol above the global minimum, respectively, considered as high in energy; thus they are not discussed in this section. Noting that the Cp− as 6 π-electron donor is bound to the scandium metal through an η5-coordination manner for all isomers.
Titanium model complexes
The optimized geometries of [(Cp)Ti]2(Phn) species show that the syn structures are more stable than those of anti ones regardless the conformation type (Fig. 3). The syn-[(Cp)Ti]2(Phn) singlet structure corresponds to the coordination of the adjacent rings is obtained as global minimum exhibiting a small HOMO-LUMO gap of 0.40 eV (Table 2), while its homolog of triplet state is not found as energy minimum structure characterized by a large imaginary frequency of − 377 cm−1, where the relative energies increase between the global minimum exhibiting a direct Ti–Ti contact and those of separate Ti centers. The syn-[(Cp)Ti]2(Phn) structure is obtained as global minimum lying lower in energy by 13.3 and 11.3 kcal/mol than the anti-[(Cp)Ti]2(Phn) singlet and triplet structures, respectively, due to the gain of Ti–Ti bonding. The syn and anti structures of the conformation (b) are found higher in energy than the global minimum whatever the spin state; therefore, they are not presented in Table 2 and Fig. 3. The Ti−C bond distances for the syn-[(Cp)Ti]2(Phn) structure in the range 2.120−2.387 (BP86) and 2.111−2.359 Å (BP86-D) are short; thus they give rise to an η6,η6 coordination mode between the phenazine ligand and the (Cp)Ti-(Cp)Ti metallic fragment. The Ti−Ti bond distance of 3.006 against 3.000 Å (BP86-D) suggests probably the presence of multiple bonds. Consequently, the formal bond order can be attributed on the basis of the bond distance and the MO localization and corroborated by the determination of the WBI value of 0.72, conducting to formal Ti–Ti bond order of 2, thereby providing the 16-MVE configuration for each Ti(I) center, wherein the 10π-electron of the adjacent C6 and C4N2-coordinated rings are formally shared equitably by both metallic centers. The metal-metal bonding corresponds to the electronic configuration (σ)2(π)2(δ)0(δ*)0(π*)0(σ*)0 matching well with a Ti–Ti double bond highlighted by the MO plots sketched in Fig. 4 showing clearly the presence of a σ and π Ti–Ti bonding. Noting that the low C s symmetry allows σ-δ and σ*-δ* mixing for the d-orbitals implied in the d–d interactions as sketched in Scheme 2. The syn and anti-[(Cp)Ti]2(Phz) quintet structures of configuration (a) lie 9.1 and 11.5 kcal/mol above the global minimum exhibiting comparable coordination modes, but with slight Ti–C and Ti–N bond distance elongations. The syn-[(Cp)Ti]2(Phz) quintet structure shows a considerable lengthening of the Ti–Ti bond distance from 3.006 to 3.397 Å and the weakening of the WBI from 0.72 to 0.35. For all Ti isomers whatever the configuration and the spin state, the Cp− ligand is connected via η5-coordination to metal.

Optimized [CpM]2(ƞ6, ƞ6–Phz) (M = Sc, Ti, V) singlet and triplet structures in their syn and anti configurations. Relative energies ∆E between isomers are given in (kcal/mol). S, T and Q indicate singlet, triplet and quintet spin states

MO diagrams of syn-[CpSc]2(ƞ6,ƞ6-Phz), syn-[CpTi]2(ƞ6,ƞ6-Phz) and syn-[CpV]2 (ƞ6, ƞ6-Phz) singlet complexes of C s symmetry

The six metallic MOs composing the bonding and antibonding d-block
The syn-[(CO)3Ti]2(Phn) and syn-[(PH3)3Ti]2(Phn) species possess two supplementary electrons than the electron-deficient syn-[(Cp)Ti]2(Phn) species should in principle result in the cancelation of the electron deficiency which should occupy a δ-bonding orbital. Evidently, it is what has happened, where the MO diagram of Fig. 2 shows clearly the occupation of δ-bonding MO, while its counterpart δ* antibonding remains empty, thus giving rise to δ Ti−Ti bond. Accordingly for the syn-[(CO)3Ti]2(Phn) (Fig. 1) and syn-[(PH3)3Ti]2(Phn) (Fig. S1) singlet C s structures exhibiting moderate HOMO-LUMO gaps of 0.40 and 0.63 eV, respectively, in which the Ti−Ti bonding is described by a formal triple bond order consistent with the bond distance of 3.023 and 3.110 Å and the WBI values of 0.86 and 0.83 (Table 1 and Table S1), respectively, conducting to both neutral Ti(0) centers of 18-MVE closed-shell configuration matching well with the electronic configuration (σ)2(π)2(δ)2(δ*)0(π*)0(σ*)0. The passage from the syn-[(CO)3Ti]2(Phn) singlet structure to the triplet and quintet ones gives rise to structures closeness in energy as gathered in Table 1. This passage leads to Ti–Ti bond distance elongation from 3.020 (singlet structure) to 3.132 (triplet structure) and 3.137 Å (quintet structure) and the fall of the WBI from 0.83 (formal bond order of 3 for the singlet structure) to 0.51 (formal bond order of 2 for the triplet structure) and to 0.51 (formal bond order of 1 for the quintet structure) due to the depopulation of the δ bonding orbital and the population by one electron of its δ* antibonding counterpart for the triplet structure and depopulation of the π bonding orbital and the population by one electron of its π* antibonding counterpart for the quintet structure consisting with the following electronic configuration: (σ)2(π)2(δ)1(δ*)1(π*)0(σ*)0 and (σ)2(π)1(δ)1(δ*)1(π*)1(σ*)0 for the triplet and quintet structures, respectively. Similar trends are observed for the [(PH3)3Ti]2(Phn) in terms of MO localizations, bond distance, and WBI value as elucidated by the structures displayed in Fig. S1 and the selected data regrouped in Table S1 of the supplementary information. Whereas, the quintet structures are found less stable at least by 20.0 kcal/mol than the global minimum, due to the occupation of Ti–Ti antibonding MOs, inducing relative instabilities.
Vanadium model complexes
The optimized geometries of [(Cp)V]2(Phn) species gave syn and anti structures of various configurations as energy minimum with singlet, triplet, and quintet states, in which the Cp− ligand is coordinated to vanadium through η5 fashion. The syn-[(Cp)V]2(Phn) (Fig. 3) is obtained as global minimum exhibiting a large HOMO-LUMO gap of 1.10 eV. This structure is found slightly more stable than its homolog of triplet and quintet ones by 3.0 and 2.9 kcal/mol, respectively, as gathered in Table 2, whereas the relative energies increase between the global minimum and other isomers of conformation (b) whatever the spin state. The V−C bond distances in the range 2.135–2.365 Å put emphasis on strong interactions, thereby giving rise to an η6,η6 coordination mode between the phenazine ligand and the (Cp)V–V(Cp) moiety. The vanadium–vanadium bond distance of 2.675 (BP86) and 2.660 Å (BP86-D) is considered as short and could correspond to a multiple bond. Indeed, the MO diagram sketched in Fig. 4 describes clearly the metal-metal bonding corresponding to the electronic configuration (σ)2(π)2(δ)2(δ)0(π)0(σ)0 matching well correlated to a V–V triple bond and consistent with the computed large HOMO-LUMO gap of 1.10 eV corresponding to the occupation of the three σ, π and δ components, and the depopulation of their antibonding counterparts consisting of the occupation of σ (HOMO-2, 37a′), π (HOMO-1, 25a′′), and δ (HOMO, 38a′) bonding orbitals and their antibonding vacant LUMO + 5, LUMO + 2, and LUMO, respectively, thus, leading to a 18-MVE (metal valence electrons) configuration for V(I) metal centers. This bond order is consistent with the Wiberg Bond Index value of 1.24. Furthermore, the dative σ V–V bond is made of a combination of dz 2 and dxy AOs of V1, the π V–V one is composed of pure metallic dxz AOs of each metal, while the δ V–V one is assured by a mixture of dx 2 -y 2 and dz 2 of V1 AOs and a combination of dz 2 and dxy AOs of V2. Accordingly, the singlet state isomers are calculated as the ground state for the vanadium structures regardless the deemed configuration. The vanadium triplet structure corresponds to the following electronic configuration type (σ)2(π)2(δ)1(δ*)1(π*)0(σ*)0 consists of V–V double bond which is less stable by 3.5 kcal/mol than the global minimum, a value which is not significant at the considered level of calculations. One can observe the lengthening of V–C bond distances going from the singlet structure to the triplet and quintet ones, while the V–V bond distance undergo a considerable lengthening (2.968 and 3.367 Å for triplet and quintet structures, respectively, against 2.675 Å for the singlet structure) (Table 2). For the quintet structure, the coordination is maintained, but with V–C and V–N bond distances lengthening as clearly mentioned in Table 2 and shown in Fig. 3, where the four unpaired electrons are exclusively localized on both vanadium metals based on the spin density values of 2.06 and 2.13. One can observe the V–V bond distance lengthening from 2.675 in syn-[(Cp)V]2(Phn) to 3.009 Å in syn-[(CO)3V]2(Phn), consisting with the occupation by two electrons of δ* antibonding orbital due to the relative richness of the tricarbonyl moiety than the cyclopentadienyl one as displayed by MO diagrams of Fig. 2. Unfortunately, the syn structure with V–V multiple bonds is not stabilized enough lying higher in energy than the structures without direct V–V bonding as gathered in Table 1. Consequently, the different syn and anti structures of conformation (b) are closeness in energy. The syn and anti singlet structures of conformations (a) and (b) exhibit small HOMO-LUMO gaps of 0.50 and 0.42 eV, respectively, in which each (CO)3V fragment is bound to one C6 ring by means of η6-coordination mode, where the phenazine is considered as dicationic ligand allowing to each metal to behave like monoanionic V(− I) center acquiring the 18-MVE configuration. For the [(PH3)3V]2(Phn) models (see Supplementary information), the anti conformation are computed more stable than those of the syn ones regardless the considered conformation (Fig. S1 and Table S1).
Chromium model complexes
Syn and anti configurations of singlet, triplet, and quintet structures are found as energy minimum for [(CO)3Cr]2(Phn) species (Fig. 5 and Table 3). The structures without direct Cr–Cr bonding are computed more stable than those of direct ones associated with the Lewis formula giving the phenazine as dianionic ligand. The (CO)3Cr fragments prefer coordinate both terminal rings rather than the adjacent ones providing singlet and triplet of the C 2v (syn) and the C 2h (anti) arrangements as lowest energy structures for which the closed-shell configuration is more stable by 17.8 and 17.5 kcal/mol than their corresponding structures of the open-shell one, respectively. The syn and anti singlet structures of conformation (b) are closeness in energy (the relative energy does not exceed 2.5 kcal/mol) displaying comparable significant HOMO-LUMO gaps of 0.96 and 1.02 eV, for which the Cr−C bond lengths are comparable (Table 3) leading to a perfect η6-coordination mode reproducing the experimental bonding for the molybdenum binuclear complexes [46]. Based on the fact that the phenazine is formally considered as dianionic ligand, thus the chromium atoms correspond to one Cr(II) and the other to Cr(0) with 16- and 18-MVE, respectively. Whereas, the structure of conformation (a) with long Cr–Cr distance of 3.349 (BP86) and 3.331 Å (BP86-D) comparable to that found for (CO)6Cr2(Az) of 3.325 Å [47] is disfavoured compared to those of conformation (b), in spite of the fact that they exhibit the similar η6,η6-coordinaton mode, doubtless, the difference resides in the behavior of the phenazine ligand, which behaves as neutral and dianionic in structures of configuration (a) and (b), respectively.

Optimized [(CO)3M]2(Phz) (M = Cr, Mn) singlet and triplet structures in their syn and anti configurations. Relative energies ∆E between isomers are given in (kcal/mol)
For the [(PH3)3Cr]2(Phn) species, the syn singlet structure is found as the global minimum (Fig. S2) responding to the similar tendencies observed for Mo complexes investigated recently by us, which are in opposite to the experimental observations for the more crowded methyl ligands used instead hydrogen atoms. The global minimum for [(PH3)3Cr]2(Phn) is an anti singlet structure of configuration (b) following similar trends than those of [(CO)3Cr]2(Phn), but exhibit short Cr–C and Cr–N bond distances compared to those obtained for [(CO)3Cr]2(Phn) as displayed in Table 3 and Table S2. It is worth mentioning that the most stable isomer among the [(CO)3Cr]2(Phn) and [(PH3)3Cr]2(Phn) quintet structures is found 55.1 and 47.5 kcal/mol above its corresponding global minimum, respectively; thus they are not considered in the discussion.
For [(Cp)Cr]2(Phn), despite the syn configuration (a) with η6-η4 coordination mode offers the possibility of a direct Cr–Cr interaction, it was revealed to be closeness in energy with those of the syn and anti structures of configuration (b) with η6-η6 coordination mode due likely to repulsive interactions between cyclopentadienyl ligands besides the difference of their coordination modes (Fig. 6 and Table 4). Indeed, the [(Cp)Cr]2(Phn) singlet structures do not correspond to energy minimum, while the quintet one is found as the global minimum lying below the triplet structures at least by 10.0 kcal/mol as summarized in Table 4 and shown in Fig. 6.

Optimized [CpCr]2(Phz) singlet, triplet, and quintet structures in their syn and anti configurations (a) and (b). Relative energies ∆E between isomers are given in (kcal/mol)
Manganese model complexes
The optimized syn and anti structures of configuration (a) of singlet and triplet states lie very high in energy than those of the conformation (b); for example, the syn-[(CO)3Mn]2(Phn) lies 24.2 kcal/mol above the global minimum (Fig. 5 and Table 3). Furthermore, the optimized [(CO)3Mn]2(Phn) quintet structures are found higher in energy than the global minimum by at least 60.0 kcal/mol and are not obtained as energy minimum characterized by large imaginary frequencies. A common feature of these structures is the η4-coordination mode of both external C6 rings of the phenazine ligand. The syn and anti singlet structures of configuration (b) are closeness in energy exhibiting large HOMO-LUMO gaps of 1.54 and 1.47 eV, respectively. For these structures, one metal is described as cationic Mn(+I) and the other is anionic Mn(− I) corresponding to 16- and 18-MVE configurations, respectively, associated with the neutral phenazine ligand. Whereas, the triplet structure show an η4-coordination mode is found higher in energy than the global minimums (25.6 kcal/mol), where both manganese are neutral Mn(0) centers consistent with the 17-MVE configuration, where the unpaired electrons are located on the metal centers evidenced by the spin density value of 0.95. These results are comparable to those of [(CO)6Mn]2(Az) in terms of stability order between isomers and geometrical parameters [48].
Similarities are obtained for [(PH3)3Mn]2(Phn) complexes, where the anti-[(PH3)3Mn]2(Phn) structures are found more stable than those of the syn-[(PH3)3Mn]2(Phn) ones as shown in Fig. S2 and Table S2. Noting that the [(PH3)3Mn]2(Phn) quintet structures whatever the considered configuration are found less stable at least by 47.2 kcal/mol than the global minimum, a value which is much important; thus the quintet high-spin structures are not discussed for Mn metal. It is interesting to note that the geometrical parameters of different structures are not sensitive to the spin state variation due to the fact that the depopulated and populated orbitals are metallic nonbonding ones.
Iron model complexes
The (Cp)Fe fragments avoid the face-to-face arrangement of the conformation (a), where the corresponding singlet and triplet structures lose energy in favor of the remote positions corresponding to the syn and anti singlet structures than those of (Cp)Fe 43.2 and 39.0 kcal/mol compared to the lowest energy syn structure of configuration (b). These lowest energy structures (Fig. 7 and Table 5) exhibit large HOMO-LUMO gaps of 1.31 and 1.34 eV showing an η4-coordination of each terminal C6 ring with short Fe–C bond distances in the range 2.060–2.129 Å. In both structures, one Fe center is considered as neutral Fe(0) and the second as dictionic Fe(II), giving rise to 18 and 16-MVE closed-shell configurations.

Optimized [CpM]2(Phz) (M = Fe, Co) singlet and triplet structures in their syn and anti configurations (a) and (b). Relative energies ∆E between isomers are given in (kcal/mol). S and T indicate singlet and triplet spin states, respectively
The optimized geometries of iron metal connected to tricarbonyls or triphosphines possessing two more electrons than those of (Cp)Fe ones adopt various structures as sketched in Fig. 8 and Fig. S3. The (PH3)3Fe and (CO)3Fe as 14 electron fragments would behave differently than the (Cp)Fe one as 13 electrons. The structures displayed in Fig. 8 show the preference of the singlet ones than those of the triplet, wherein the structures corresponding to the coordination of both terminal C6 rings are more stable than those coordinating the adjacent ones of both syn and anti configurations. The syn-(CO)6Fe2(Phn) and anti-(CO)6Fe2(Phn) singlet C s structures of configuration (b) are obtained 4.0 kcal/mol below than their corresponding triplet structures as summarized in Table 6. Whereas, the syn and anti in which the metallic fragment coordinating the adjacent rings are found high in energy compared to those of conformation (b). The syn-(CO)6Fe2(Phn) and anti-(CO)6Fe2(Phn) structures display large HOMO-LUMO gaps of 1.04 and 1.02 eV, respectively, wherein each terminal ring is connected to each (CO)3Fe through η4-coordination manner, thus each Fe(0) center attains the 18-MVE configuration. This type of coordination mode gives rise to a twisted phanazine ligand accompanied by shift of each (CO)3Fe towards the external carbon atoms. Comparable tendencies are observed for the (PH3)3 in terms of coordination and electronic configuration. The low-spin/high-spin splitting energy is reduced. Surprisingly, the geometry optimizations without symmetry constraints of the syn configuration (a) gave a distorted structure with a direct Fe–Fe bonding (Fig. 8), wherein the Fe2 metal center is coordinated to the C4N2 ring via the lone pair of the nitrogen atom, while the Fe1 is in η4-coordination mode with the C6 ring and one carbonyl among six is bridging ligand is obtained as the global minimum lying about 7.5 kcal/mol below described above. For this unexpected structure, both iron centers satisfy the 18-MVE rule considering a Fe–Fe single bond distance of 2.580 Å corroborated by a WBI of 0.22, which exhibits large HOMO-LUMO gap of 1.30 eV. The MOs sketched in Scheme 3 show that the HOMO-9 corresponds to the σ Fe-Fe bonding orbital; the HOMO is Fe–N bonding, while the LUMO is its antibonding counterpart, thus occupying this orbital would broke the corresponding bond as elucidated in Scheme 3. Indeed, it is what has happened for the triplet structure, where one electron is located in this LUMO, giving rise to a triplet and quintet structures less stable than the singlet one by 3.1 and 20.2 kcal/mol, characterized by a decrease of the Fe–Fe bond length from 2.580 of the singlet structure to 2.543 and to 2.556 Å of triplet and quintet ones, respectively, but connected to the C4N2 ring via the nitrogen atom as sketched in Fig. 8. Same tendencies have been obtained for the investigated Fe2(CO)6(Az), where the syn singlet structure has been found as the global minimum [49].

Optimized [(CO)3M]2(Phz) (M = Fe, Co) singlet and triplet structures in their syn and anti configurations. Relative energies ∆E between isomers are given in (kcal/mol)

HOMO (a), HOMO-9 (b), and LUMO (c) for [(CO)3Fe2](Phn) and HOMO (d) and LUMO (e) for [(CO)3Co2](Phn)
Cobalt model complexes
The anti-[(CO)3Co]2(Phn) structures of singlet and triplet states are indistinguishable (Fig. 8) lying on plate potential energy hypersurface as clearly regrouped in Table 6. Surprisingly, all the optimized geometries show only an η2-coordination mode independently of the considered conformation giving rise to deficient cobalt Co(I) centers of 16-MVE, in accordance with the vacant non-bonding LUMO and LUMO + 1 molecular orbitals, which are purely metallic ones (Scheme 3). However, The syn-[(CO)3Co]2(Phn) triplet structure optimized without constraints of symmetry was revealed to be the more stable isomer, exhibiting a distorted arrangement. This global minimum displays a direct metal-metal bonding of 2.484 with WBI value of 0.29, where Co is not in coordination with the central C4N2 ring. The spin density values of 0.12 and 1.74 show that the unpaired electrons are mostly localized on only one metal center rather than on both. It is worth noting that the syn-[(CO)3Co]2(Phn) quintet C 1 structure lies 40.1 kcal/mol above the triplet one as the global minimum and displays a distorted arrangement, where the Co metal is not connected to the C4N2 ring.
With two electrons less than [(CO)3Co]2(Phn), the [(Cp)Co]2(Phn) complexes (Fig. 7) show structures with η4-coordination mode instead of η2 one conducting to both cationic Co(I) centers satisfying the 18-electron rule resembling to the isoelectronic [(CO)3Fe]2(Phn) ones and behaving like as syn-[Cp(Co)]2(η4,η4-Az) [50]. The different closed shell structures exhibit large HOMO-LUMO gaps at least of 0.94 eV. The Co–C bond distances within the range putting emphasis on strong interactions.[51,52,53]
Nickel model complexes
All [(Cp)Ni]2(Phn) structures found as an energy minimum (Fig. 9) show an η2-coordination mode. The syn-[(Cp)Ni]2(Phn) and anti-[(Cp)Ni]2(Phn) triplet structures, where each (Cp)Ni fragment is bound to one terminal C6 ring, are closeness in energy and are obtained as global minimums giving rise to both cationic Ni(I) centers with 17-MVE each corresponds to the localization of the two unpaired electrons on the metal centers as evidenced by the spin density values of 0.98. Indeed, the corresponding singlet structures lie 5.0 kcal/mol above those of the triplet ones corresponding to Ni(II) and Ni(0) centers of 16- and 18-MVE, respectively. It is worth mentioning that the [(CO)3Ni]2(Phn) and [(PH3)3Ni]2(Phn) having two supplementary electrons than those of [(CO)3Co]2(Phn) display uncoordination between (L)3Ni (L = CO, PH3) and phenazine moieties, whose Ni–C distances are more than 2.35 Å, which are beyond the range of the corresponding bond distances. The nickel high-quintet structures are found very high in energy compared to those of the low spin ones.

Optimized [CpNi]2(Phz) singlet and triplet structures in their syn and anti configurations. Relative energies ∆E between isomers are given in (kcal/mol). S and T indicate singlet and triplet spin states, respectively
Conclusion
This study reports a theoretical investigation of the electronic and the molecular structure of [(L3M)2](Phz) complexes for first-row transition metals (Sc–Ni) coordinated to the phenazine ligand in their syn and anti conformations. The syn conformation offers the possibility of a direct metal-metal interaction.
We have shown that the electronic communication between the metal centers depends on the nature of the metal and governed by the behavior of the phenazine ligand. The C–C and C–N in the coordinated rings undergo some modifications, indicative of donation and backdonation of the (CO)3M, (PH3)3M, and CpM fragments to the phenazine π-MOs. Thus, the coordination destroys the planarity of the phenazine ligand.
We have shown that most of the investigated compounds should be enough “stable” for being isolated. These results show the capability of the phenazine ligand to adapt itself to the electronic demand of the metals, in agreement with the nature of the metal-ligand bonding. The metal-metal bonding decreases with the increasing of the metal valence electron count, where the chromium binuclear complexes are intermediate between the presence and the absence for such bonding. Indeed, single, double, and triple bonds are suggested for Sc, Ti, and V complexes, respectively. Our findings showed that the iron and cobalt avoid the symmetrical structures and adopt distorted ones with the formation of Fe–Fe and Co–Co single bonds. Among all optimized structures, only the high-quintet anti-[(Cp)Cr]2(Phz) structure is found as the global minimum; however, the scandium, manganese, iron, cobalt, and nickel quintet structures are disfavoured compared to those of low spin and those of titanium; vanadium are closeness in energy with their homologs of singlet and triplet ones.
References
Kallir AJ, Suter GW, Wild UP (1985). J Phys Chem 89:1996
Aaron JJ, Maafi M, Parkanyi C, Boniface C (1995). Spectrochim Acta 51A:603
Hirata Y, Tanaka I (1976). Chem Phys Lett 43:568
Pavlopoulos TG (1987). Spectrochim Acta 43A:715
Del Barrio JI, Rebato JR, Tablas FMG (1989). J Phys Chem 93:6836
Kuzmin VA, Levin PP (1988). Bull Acad Sci USSR Div Chem Sci 37:1098
Casey CP, Audett JD (1986). Chem Rev 86:339
Schwab PFH, Levin MD, Michl J (1999). Chem Rev 99:1863
Holton J, Lappert MF, Pearce R, Yarrow PI (1983). Chem Rev 83:135
Moss JR, Scott LG (1984). Coord Chem Rev 60:171
Chowdhury MDAH, Rahman MDS, Islam MDR, Rajbangshi S, Ghosh S, Hogarth G, Tocher DA, Yang L, Richmond MG, Kabir SE (2016). J Organomet Chem 805:34
Shuster V, Gambarotta S, Nikiforov GB, Budzelaar P (2013). Organometallics 32:2329
Zhu G, Tanski JM, Churchill DG, Janak KE, Parkin G (2002). J Am Chem Soc 124:13658
Zhu G, Tanski JM, Parkin G (2003). Polyhedron 22:199
Zhu G, Pang K, Parkin G (2008). J Am Chem Soc 130:1564
Zhu G, Pang K, Parkin G (2008). Inorg Chim Acta 361:3221
Zendaoui MS, Zouchoune B (2013). Polyhedron 51:123
Merzoug M, Zouchoune B (2014). J Organometal Chem 770:69
Zouchoune F, Zendaoui S-M, Bouchakri N, Djedouani A, Zouchoune B (2010). J Mol Struct 945:78
Farah S, Korichi H, Zendaoui SM, Saillard JY, Zouchoune B (2009). Inorg Chim Acta 362:3541
Bensalem N, Zouchoune B (2016). Struct Chem 27:1781
Fadli S, Zouchoune B (2016). Struct Chem 28:985
Zendaoui SM, Saillard JY, Zouchoune B (2016). Chem Select 5:940
Saiad A, Zouchoune B (2015). Can J Chem 93:1096
Zendaoui MS, Zouchoune B (2016). New J Chem 40:2554
Benhamada N, Bouchene R, Bouacida S, Zouchoune B (2015). Polyhedron 91:59
Chekkal F, Zendaoui SM, Saillard JY, Zouchoune B (2013). New J Chem 37:2293
Stone AJ (1997) The theory of intermolecular forces. Oxford University Press, Oxford
Kaplan IG (2006) Intermolecular interactions. Wiley, Chichester
Grimme S (2006). J Comput Chem 27:1787
SCM ADF2014.01, theoretical chemistry. Vrije Universiteit, Amsterdam
Baerends EJ, Ellis DE, Ros P (1973). Chem Phys 2:41
te Velde G, Baerends EJ (1992). J Comput Phys 99:84
Fonseca Guerra C, Snijders JG, te Velde G, Baerends EJ (1998). Theo Chim Acc 99:391
Bickelhaupt FM, Baerends EJ (2000). Rev Comput Chem 15:1
te Velde G, Bickelhaupt FM, Fonseca Guerra C, van Gisbergen SJA, Baerends EJ, Snijders JG, Ziegler T (2001). J Comput Chem 22:931
Vosko SD, Wilk L, Nusair M (1990). Can J Chem 58:1200
Becke AD (1986). J Chem Phys 84:4524
Becke AD (1988). Phys Rev A 38:3098
Perdew JP (1986). Phys Rev B 33:8822
Perdew JP (1986). Phys Rev B 34:7406
Versluis L, Ziegler T (1988). J Chem Phys 88:322
Fan L, Ziegler T (1992). J Chem Phys 96:9005
Fan L, Ziegler T (1992). J Phys Chem 96:6937
P. Flükiger, H. P. Lüthi, S. Portmann, J. Weber, MOLEKEL, Version 4.3.win32 Swiss Center for Scientific Computing (CSCS), Switzerland, 2000–2001. http://www.cscs.ch/molekel/
Wiberg KB (1968). Tetrahedron 24:1083
Weinhold F, Landis CR (2005) Valency and bonding: a natural bond order donor acceptor perspective. Cambridge University Press, Cambridge
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Landis CR, Weinhold F (2013) NBO 6.0.; Theoretical chemistry institute. University of Wisconsin, Madison Available at: www.chem.wisc.edu/_nbo6. Accessed 1 Feb 2013
Sattler A, Zhu G, Parkin G (2008). J Am Chem Soc 131:3221
Wang H, Sun Z, Xie Y, King RB, Schaefer HF III (2010) Eur J Inorg Chem: 5161
Sun Z, Wang H, Xie Y, King RB, Schaefer III HF (2010). Dalton Trans 39:10702
Wang H, Sun Z, Xie Y, King RB, Schaefer III HF (2010). Organometallics 29:630
Korichi H, Zouchoune F, Zendaoui SM, Zouchoune B, Saillard JY (2010). Organometallics 29:1693
Acknowledgements
The authors are grateful to the Algerian MESRS (Ministère de l’Enseignement Supérieur et de la Recherche Scientifique) and DGRSDT (Direction Générale de la Recherche Scientifique et du Développement Technologique) for the Financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
Selected geometrical and energetic parameters calculated for [(PH3)3M]2(Phz) (M = Sc, Ti, V, Cr, Mn, Fe, and Co) models obtained by BP86 method of singlet (S = 0) and triplet (S = 1) spin states and various symmetries (Tables S1–S3) and optimized-BP86 [(PH3)3M]2(Phz) (M = Sc, Ti, V, Cr, Mn, Fe, and Co) structures of of singlet (S = 0) and triplet (S = 1) spin states for syn and anti conformations (Figs. S1–S3) (DOCX 1698 kb)
Rights and permissions
About this article

Cite this article
Naili, N., Zouchoune, B. Structural diversity of homobinuclear transition metal complexes of the phenazine ligand: theoretical investigation. Struct Chem 29, 725–739 (2018). https://doi.org/10.1007/s11224-017-1064-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-017-1064-2