
Abstract
The reaction of N-benzoylphosphoramidic dichloride with amines afforded some new N-benzoylphos-phoric triamides with formula C6H5C(O)NHP(O)(X)2, X=NH–CH(CH3)2 (1), NH–CH2–CH(CH3)2 (2), NH–CH2–CH(OCH3)2 (3), N(CH3)[CH2CH(OCH3)2] (4) and N(CH3)(C6H11) (5) that were characterized by 1H,13C,31P NMR, IR spectroscopy and elemental analysis. The structures have been determined for compounds 4 and 5 by X-ray crystallography. These compounds contain one amidic hydrogen atom and form centrosymmetric dimmers via intermolecular –P–O⋯H–N–hydrogen bonds besides weak C–H⋯O hydrogen bonds that lead to three-dimensional polymeric clusters in the crystalline lattice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, investigations on N-benzoylphosphoric triamides are an important part of phosphoramidate chemistry due to their synthetic and structural [1–3], coordination [4–8] and biological [9] applications. These molecules have –C(O)NHP(O)– skeleton and the existence of peptide group in these derivatives cause them biologically active anticancer drugs [10]. Moreover, applications of other phosphoramidates in chemical reactions have been reported [11–13]. Compound C6H5C(O)NHP(O)[NH–C(CH3)3]2 showed two conformers in solution NMR spectra and solid state [14]. Moreover, compounds C6H5C(O)NHP(O)X2, X = NC3H6 [15], NC4H8 [16] 4-CH3–C6H4OP(O)(NH–CH(CH3)(CH2C6H5) [17] and C6H5C(O)NHP(O)(NC5H10)2 [18] 4-F-C6H4C(O)NH P(O)(NC5H10)2 [19] exhibited two and four conformers, respectively, only in their crystalline states. The synthesis and structure of compound {(1R, 2S)-N-iPr-ephedrine}P(S)NH[(Si(CH3)3], which is an oxazaphosphole molecule with three independent molecules in the crystalline lattice, was reported [20]. The spectra and structure of some organophosphorus compounds (that are analogues of phosphoramidates) [21] and also of a polymorph Mo2(CO)8(μ-PPh2)2 complex have been investigated [22]. Herein, we have considered the synthesis and spectroscopic characterization of several new N-benzoylphosphoric triamides with formula C6H5C(O)NHP(O)(X)2, X=NH–CH(CH3)2 (1), NH–CH2–CH(CH3)2 (2), NH–CH2–CH(OCH3)2 (3), N(CH3)[CH2CH(OCH3)2] (4) and N(CH3)(C6H11) (5). The solid-state crystal structures of compounds 4 and 5 were investigated by means of X-ray crystallography technique.
Experimental
Crystal structure determination
X-ray data of compounds 4 and 5 were collected on a Bruker SMART 1000 CCD area detector [23] single crystal diffractometer with graphite monochromated Mo Kα radiation (λ=0.71073 Å). The structures were refined with SHELXL-97 [24] by full-matrix least-squares on F 2. The positions of hydrogen atoms were obtained from the difference Fourier map. Routine Lorentz and polarization corrections were applied and an absorption correction was performed using the SADABS program [25].
Spectroscopic measurements
All reactions were performed under argon atmosphere and in dry solvents. 1H, 13C and 31P NMR spectra were recorded on a Bruker Avance DRS 500 spectrometer. 1H and 13C chemical shifts were determined relative to internal TMS, 31P chemical shifts relative to 85% H 3PO4 as external standard. Infrared (IR) spectra were recorded on a Shimadzu model IR-60 spectrometer. Elemental analysis was performed using a Heraeus CHN-O-RAPID apparatus. C 6H5C(O)NHP(O)Cl2 was prepared according to the literature method [26].

Synthesis pathway of compounds 1–5
Results and discussion
Spectroscopic study
Synthesis of compounds 1–5 was performed by the reaction of N-benzoylphosphoramidic dichloride [26] with corresponding amines, Scheme 1. The spectroscopic data of these molecules are summarized in Table 1. 1H NMR spectra of compounds 1, 2 and also of 4 indicate two separate signals for the two non-equivalent CH3 and OCH3 groups, respectively. In these compounds, the existence of prochiral CH group leads to different methyl and methoxy groups. 2 J(PNH)amine coupling constant for amine protons in compound 1 is 9.1 Hz in d6-DMSO and 13.2 Hz in CDCl3. This constant in both similar compounds 2 (containing isobutyl groups) and 3 (containing 2,2-dimethoxy-ethyl groups) is 16.1 Hz that is higher than those observed for acyclic phosphoramidates [14, 27, 28]. 3 J(PNCH) coupling constant in molecule 4 is 16.3 Hz, which is higher than the observed values for our previously reported acyclic phosphoramidates [27–29]. This constant is 10.9 Hz in molecule 5 (for the splitting of N-methyl protons with phosphorus atom). The CH2 protons in compound 4 are diastereotopic and exhibit two ddd coupling patterns in 1H NMR spectrum due to the coupling of each of these protons with another one, with CH proton and phosphorus atom. The amidic proton splits with phosphorus atom in compounds 1 and 5 with 2 J(PNH)amide = 5.7 and 5.9 Hz, respectively.
13C NMR spectra of compounds 1 and 2 display two distinct signals for the two non-equivalent methyl carbon atoms. The related 3 J(P, C) coupling constants for the methyl carbon atoms in 1 are 4.8 and 6.6 Hz. The ipso carbon atoms of phenyl rings in compounds 1–5 show 3 J(P, C)aromatic coupling constants in the range of 7.5 Hz (in 3) to 8.7 Hz (in 4). The coupling between C–O carbon atom and phosphorus atom have been observed only in compound 5 (2.9 Hz). The only difference between compounds 3 and 4 is the replacement of amino proton in NH–CH2CH(OCH3)2 chains (3) with CH3 group (4). A comparison between compounds 3 and 4 indicates that the 3 J(P, C)aliphatic for the splitting of carbon atom in CH moiety with phosphorus atom is nearly twice in 3 (6.7 Hz) relative to that of in 4 (3.1 Hz). The δ(31P) of compounds 1–5 are in the range of 13.75 ppm (in 5) to 8.22 ppm (in 1).
IR spectra of compounds 1–5 showed that the ν P–O and ν C–O frequencies are in the range of 1182 cm−1 (in 4) to 1215 cm−1 (in 2) and 1635 cm−1 (2) to 1670 cm−1 (5), respectively. The P–O bond frequency in compound 2 (containing isobutyl substituent) is stronger than in 1 (containing isopropyl substituent), but for the C–O frequency an opposite result was obtained. A comparison between similar compounds 2 and 3 indicates that ν P–O in 2 is stronger than in 3 in contrary to the result for ν C–O. This is also observed for compounds 3 and 4 in which the ν P–O in 3 is stronger than in 4. The ν P–O in compounds 4 and 5, 1182 and 1183 cm−1, are very close to each other that shows the P–O bond lengths in these molecules are nearly the same. This is confirmed by X-ray crystal structures of 4 and 5 indicate that the P–O bond lengths are 1.4818(11) and 1.4842(12) Å, respectively.

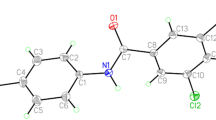
Molecular structure and atom labelling scheme for compound 4 (50% probability ellipsoids)

The two non-equivalent N(CH3)[CH2CH(OCH3)2] groups in structure of 4 that indicates different spatial orientations of N-methyl groups and two OCH3 moieties
X-ray crystallography investigation
Single crystals of compound 4 were obtained from a mixture of diethylether/n-heptane and of 5 from a mixture of dichloromethane/n-hexane/diethylether at room temperature. The crystal data and the details of the X-ray analysis are given in Table 2, selected bond lengths and angles in Table 3. Molecular structures (ortep view) of these compounds are shown in Figs. 1 and 4, the unit cells are presented in Figs. 3 and 6.

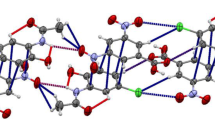
A view of the unit cell packing of compound 4

Molecular structure and atom labelling scheme for compound 5 (50% probability ellipsoids)

The two non-equivalent N(CH3)(C6H11) groups in compound 5 that indicates different spatial orientations of N-methyl groups and two cyclohexyl moieties
As we observed in NMR section, the two methoxy groups of one chain in molecule 4 are not equivalent with each other due to their different spatial orientations. This matter can be attributed to the existence of prochiral CH group in this compound and it can be shown by the differences in their similar torsion angles. The torsion angles O(1)–P(1)–N(2)–C(9) and O(1)–P(1)–N(3)–C(14) of the two amine chains are 114.01(14)° and −34.21(14)°, respectively, (similarly, compare the torsion angles P(1)–N(2)–C(9)–C(10) and P(1)–N(3)–C(14)–C(15) that are −87.10(16)° and 97.12(15)°, respectively). The two N-methyl groups have different orientations relative to each other that can be displayed by the differences in their related torsion angles. The torsion angles O(1)–P(1)–N(2)–C(8) and O(1)–P(1)–N(3)–C(13) are −61.38(15)° and 158.33(13)°, respectively, Fig. 2. Also, in compound 5, the two N-methylcyclohexyl moieties are not equivalent (Fig. 5) and their similar torsion angles are different. The torsion angles O(1)–P(1)–N(2)–C(8) and O(1)–P(1)–N(3)–C(15) are −63.86(14)° and 167.69(12)°, respectively. Moreover, the torsion angles O(1)–P(1)–N(2)–C(9), O(1)–P(1)–N(3)–C(16) and N(3)–P(1)–N(2)–C(8), N(2)–P(1)–N(3)–C(15) are 93.85(13)°, 167.69(12)° and 170.43(12)°, −63.27(14)°, respectively.
In molecules 4 and 5 the phosphoryl and the carbonyl groups show anti configuration, Figs. 1 and 4. The phosphorus atoms in these structures have distorted tetrahedral configuration. The bond angles around P(1) atoms in these compounds are in the range of 105.14(7)° to 117.30(7)° (both of them have been observed in 5), for the angles N(2)–P(1)–N(1) and O(1)–P(1)–N(2), respectively, Table 3. In compounds 4 and 5, the angles OPNamide (Namide is the nitrogen atom of P(O)N(H)C(O) moiety) are lower than the angles OPNamine (Namine is the nitrogen atom of P(O)NR moiety). This was also observed in our previously reported compounds [28, 29, 31]. The P=O bond lengths in molecules 4 and 5 are 1.4818(11) and 1.4842(12) Å that are larger than the normal P–O bond length (1.45 Å) [30].

The unit cell packing of molecule 5 (intermolecular hydrogen bonds are shown by dashed lines)
Compounds 4 and 5 each contain one amidic hydrogen atom and form centrosymmetric dimmers via intermolecular –P–O⋯H–N– hydrogen bonds (Table 4). Considering weak C–H⋯O hydrogen bonds leads to three-dimensional polymeric clusters in the crystalline network of these molecules. The earlier studied N-benzoyl- and N-4-fluorobenzoylphosphoric triamides exist either in the form of dimmeric aggregates [15, 19, 29] or as polymeric chains [28, 31].
The P– Namide bond lengths are longer than the P–Namine bond lengths, because of the resonance interaction of the Namide with the C–O π system that cause a partial multiple bond character in C–Namide (the C–Namide bond lengths are shorter than the C–Namine bond lengths, Table 3). All of these P–N bonds are shorter than the typical P–N single bond length (1.77 Å [30]). This is probably due to the electrostatic effects (polar bonds) which overlap with P–N σ bond [32]. The P–O, P–Namine and P–Namide bond lengths in compound 5 are slightly longer than in 4. These bond lengths in our previously reported structures were in the range of 1.406(12) Å (in 8) to 1.488(1) Å (in 9), 1.527(12) Å (in 10) to 1.658(11) Å (in 10), and 1.661(13) Å (in 8) to 1.747(9) Å (in 10), respectively (Table 5).The environment of the nitrogen atoms is practically planar. In compound 5, the angles C(8)–N(2)–C(9), C(8)–N(2)–P(1) and C(9)–N(2)–P(1) are 117.26(13)°, 117.50(11)° and 121.53(11)°, respectively with average 118.8°. The sum of surrounding angles around N(1) and N(3) atoms are 360.08° and 356.21°, respectively. Similar results were obtained for the nitrogen atoms of structure 4 that confirm the sp2 hybridization for the N atoms, although due to the repulsion and steric interactions, some angles are greater, and the others are smaller than 120°.
Conclusion
The NMR spectra of compounds 1–5 indicated some interesting points that are due to the existence of prochiral carbon atoms (in 1–4). In compound 5, the different spatial orientations of N-methyl and cyclohexyl groups of the two chains cause asymmetry in this compound. 1H NMR spectra of compounds 1, 2 and 4 indicate two separate signals for the two non-equivalent CH3 and OCH3 groups, respectively. Crystal structures of molecules 4 (and 5) showed that the two methoxy groups (and the two N-methylcyclohexyl moieties) are not equivalent with each other due to their different spatial orientations.
Supplementary data
Spectroscopic data of compounds 1–5 can be found online as Supplementary material. Crystallographic data for the structures 4 and 5 have been deposited with Cambridge Crystallographic Data Center as supplementary publication nos. CCDC 292744 (C17H30N3O6P) and CCDC 292754 (C21H34N3O2P). Copies of the data may be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033; E-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
References
Gubina KE, Ovchynnikov VA, Amirkhanov VM, Silva TYu, Skopenko VV, Glowiak T, Kozlowski H (1999) Z Naturforschung 54B:57
Gubina KE, Ovchynnikov VA, Amirkhanov VM, Skopenko VV, Shishkin OV (1999) Z Naturforschung 54B:1357
Gubina KE, Amirkhanov VM (2000) Z Naturforschung 55B:1015
Gubina KE, Ovchynnikov VA, Swiatek-Kozlowska J, Amirkhanov VM, Silva TYu, Domasevitch KV (2002) Polyhedron 21:963
Gubina KE, Shatrava JA, Ovchynnikov VA, Amirkhanov VM (2000) Polyhedron 19:2203
Trush VA, Domasevitch KV, Amirkhanov VM, Sieler J (1999) Z Naturforschung 54B:451
Ovchynnikov VA, Timoshenko TP, Amirkhanov VM, Sieler J, Skopenko VV (2000) Z Naturforschung 55B:262
Amirkhanov VM, Ovchynnikov VA, Glowiak T, Kozlowski H (1997) Z Naturforschung 52B:1331
Amirkhanov VM, Ovchynnikov VA, Legendziewiez J, Graczyk A, Hanuza J, Macalik L (1996) Acta Physica Polonica 90:455
Riebrova ON, Biushkin WN, Malinovskij TI, Procenko LD, Dneprova TN (1982) Dokl A N USSR 266:1391
Chivers T, Krahn M, Schatte G, Parvez M (2003) Inorg Chem 42:3994
Armstrong A, Chivers T, Krahn M, Parvez M, Schatte G (2002) Chem Commun 2332
Chivers T, Krahn M, Parvez M, Schatte G (2001) Chem Commun 1992
Gholivand K, Pourayoubi M (2004) Z Anorg Allg Chem 630:1330
Gholivand K, Hosseini Z, Pourayoubi M, Shariatinia Z (2005) Z Anorg Allg Chem 631:3074
Gholivand K, Pourayoubi M, Mostaanzadeh H (2004) Anal Sci 20:51
Gholivand K, Shariatinia Z, Pourayoubi M (2005) Z Naturforschung 60B:67
Gholivand K, Vedova COD, Anaraki Firooz A, Madani Alizadehgan A, Michelini MC, Pis Diez R (2005) J Mol Struct 750:64
Gholivand K, Shariatinia Z, Pourayoubi M (2006) Polyhedron 25:711
Cain MJ, Cawley A, Sum V, Brown D, Thornton-Pett M, Kee TP (2003) Inorg Chim Acta 345:154
Chatterjee KK, Durig JR (1994) Struct Chem 5:239
Planinić P, Čalogović DM (2001) Struct Chem 12:439
Bruker (1998) SMART. Bruker Molecular Analysis Research Tool, version 5.059. Bruker AXS, Madison, WI, USA
Sheldrick GM (1998) SHELXTL version 5.10, Structure Determination Software Suit. Bruker AXS, Madison, WI, USA
Sheldrick GM (1998) SADABS version 2.01, Bruker/Siemens Area Detector Absorption Correction Program. Bruker AXS, Madison, WI, USA
Kirsanov AV, Makitra R (1956) J Gen Chem 26:907
Gholivand K, Ghadimi S, Forouzanfar A, Naderimanesh H (2001) Magn Reson Chem 39:684
Gholivand K, Shariatinia Z, Pourayoubi M (2005) Z Anorg Allg Chem 631:961
Gholivand K, Pourayoubi M, Shariatinia Z, Mostaanzadeh H (2005) Polyhedron 24:655
Corbridge DEC (1995) Phosphorus, an outline of its chemistry, biochemistry and technology, 5th edn. Elsevier, The Netherlands, pp 55–57
Gholivand K, Shariatinia Z, Pourayoubi M (2006) Z Anorg Allg Chem 632:160
Gilheany DG (1994) Chem Rev 94:1339
Acknowledgements
We wish to thank Research Council of Tarbiat Modares University for financial support of this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gholivand, K., Shariatinia, Z. Syntheses, spectroscopic study and crystal structures of some new N-benzoylphosphoric triamides. Struct Chem 18, 95–102 (2007). https://doi.org/10.1007/s11224-006-9132-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-006-9132-z