
Abstract
It was found that nano-SiO2–H2SO4 was catalyzed by the three-component cyclocondensation of aryl/heteroaryl aldehydes, hydroxylamine hydrochloride, and β-ketoesters toward the synthesis of α,β-unsaturated isoxazol-5(4H)-ones under green conditions. The reaction yielded the corresponding heterocycles at room temperature in relatively shorter reaction times. It merits mentioning that the mild conditions allow the synthesis of several α,β-unsaturated isoxazol-5(4H)-ones using this method. In this study, some new derivatives of isoxazolones were also synthesized and characterized. It is efficient, clean, simple, safe, and ecologically friendly. This straightforward method is cost-effective and requires no preparation of reactants. The three-component annulation was performed without using energy sources, for example, heat, ultrasound wave, and microwave irradiation.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multicomponent reactions (MCRs) are effective methods to the practical construction of a wide range of heterocyclic compounds in single operation with structural diversity and complexity from simple and inexpensive starting materials via the generation of several bonds in a single synthetic operation. Recently, significant consideration has been attentive to MCRs due to their simplicity, the diminished generation of the waste, shorter reaction times, step/atom economy, reduced steps of purification of intermediates, mild reaction conditions, as well as environmental benignity [1,2,3,4,5,6,7].
The isoxazol-5(4H)-one, also called isoxazolone, ring systems represent important molecular structures, which have been employed as the precursors in the synthesis of interesting organic molecules [8, 9]. They have been known to show anti-bacterial [10], antifungal [10,11,12], tyrosinase inhibitory [13], anticancer [14,15,16], anti-obesity [17, 18], anti-androgen [19, 20], CDP-ME kinase inhibitor [21], anti-HIV-1 [22], fungicide [23], and insecticides [24, 25] activities. Also, the isoxazol-5(4H)-one motifs as the powerful electron acceptor are likely to be good candidates for organic nonlinear optical (NLO) materials [26,27,28], photonic applications [29,30,31,32], and solar cells [33]. Some of the α,β-unsaturated isoxazol-5(4H)-one derivatives that show strong biological activities are presented in Fig. 1.

Selected examples of α,β-unsaturated isoxazol-5(4H)-ones with biological activities
As mentioned above, α,β-unsaturated isoxazol-5(4H)-ones play an important role in the organic synthetic chemistry. Therefore, organic chemists are interested in the synthesis of such heterocycles in recent years. One of the most attractive methods of obtaining isoxazol-5(4H)-ones is the cyclocondensation of hydroxylamine hydrochloride with β-ketoesters and various aldehydes in the presence of various types of catalysts, including sodium acetate and tungsten lamp [34], potassium phthalimide (PPI) [35], potassium hydrogen phthalate (KHP) [36], tetrabutylammonium perchlorate (TBAP)/glycine/sodium oxalate [37], 2-hydroxy-5-sulfobenzoic acid (2-HSBA) [38], N-bromosuccinimide (NBS) [39], Ag/SiO2 [40], fruit juice of citrus Limon [41], amine-modified montmorillonite nanoclay [42], sodium benzoate [12, 43], boric acid [44], SnII montmorillonite [45], nickel (II) acetate [46], citric acid [47], starch solution [48], sulfated polyborate [49], pyridinium p-toluenesulfonate (PPTS) [50], DABCO-functionalized dicationic ionic liquid (DDIL) [51], Na2S·9H2O [52], nano-MgO [53], pyridine [54,55,56], sulfanilic acid [57], KI [58], sodium silicate [59], deep eutectic solvent [60], mesolite [61], cerium chloride heptahydrate [62], phthalimide-N-oxyl [63], antimony trichloride [64], DOWEX1-x8OH [65], LiBr [66], and salicylic acid [67]. In the recent years, sulfuric acid supported on silica (SiO2–H2SO4) as an efficient and environmental-friendly solid acid catalyst has been applied in organic transformations due to their high catalytic activity, the operational simplicity, cost-effectiveness, and the recyclability. Using this convenient, safe, and highly efficient catalyst will make the products formed in high yields as well as facilitating the product separation and purification processes [68,69,70,71,72,73]. In the present study, an expeditious, one-pot, three-component cyclocondensation has been used for the green synthesis of derivatives of α,β-unsaturated isoxazol-5(4H)-ones (4a–4aq) applying nano-SiO2–H2SO4 as the catalyst under mild conditions (Scheme 1). In this contribution, the synthesis of fifteen derivatives of isoxazol-5(4H)-one is also presented for the first time.

Three-component synthesis of α,β-unsaturated isoxazol-5(4H)-ones (4a–4aq)
Experimental
General
All chemicals were purchased from Alfa Aesar and Aldrich and were used without further purification, with the exception of liquid aldehydes which were distilled and purified before using. All solvents were distilled before using. Melting points were measured on a Buchi 510 melting point apparatus and are uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded at an ambient temperature on a Bruker Avance DRX-300 MHz using DMSO-d6 as the solvent. FT-IR spectra were recorded on a PerkinElmer RXI spectrometer. The development of reactions was monitored by thin-layer chromatography (TLC) analysis on Merck pre-coated silica gel 60 F254 aluminum sheets, visualized by UV light. Elemental microanalyses were performed on an Elementar Vario EL III analyzer. SEM images were taken using a JEOL JSM-5300 microscope at acceleration voltage of 10 kV. Sample powder was deposited on a carbon tape before mounting on a sample holder. To reduce the charge developed on the sample, gold sputtering was done for 3 min.
General procedure for the synthesis of α,β-unsaturated isoxazol-5(4H)-ones (4a–4aq)
A mixture of hydroxylamine hydrochloride 2 (0.0695 g, 1 mmol), β-ketoester 3 (1 mmol), and catalyst (0.05 g) in 5 mL of distilled water was stirred at room temperature (rt) for 10 min; then, aryl/heteroaryl aldehyde 1 (1 mmol) was added to the vessel reaction. The reaction mixture was stirred at rt until the reaction was completed (monitored by TLC analysis). After the completion of the reaction, the precipitate was separated by simple filtration, washed with cold distilled water, and dried in the air. Crude products were dissolved in hot ethanol and filtered off for the separation of catalyst. The products were crystallized from ethanol (95%) to afford the title pure compounds. Spectral data for some compounds are as follows:
4-(2-Hydroxy-3-methoxybenzylidene)-3-methylisoxazol-5(4H)-one (4i)
1H NMR (300 MHz, DMSO-d6): δ = 2.25 (s, 3H, CH3), 3.85 (s, 3H, CH3O), 6.88 (t, J = 8.1 Hz, 1H, Ar–H), 7.25 (dd, J = 1.2, 8.1, 1H, Ar–H), 8.10 (s, 1H, ArCH═), 8.31 (dd, J = 1.2, 8.4 Hz, 1H, Ar–H), 10.35 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6): δ = 27.4, 75.4, 116.7, 118.7, 119.7, 139.4, 144.9, 147.7, 149.4, 152.8, 162.1, 176.0; IR (KBr, cm−1): ν = 3418, 1750, 1620, 1357, 1257, 1095, 935.
3-Methyl-4-(3,4,5-trimethoxybenzylidene)isoxazol-5(4H)-one (4k)
1H NMR (CDCl3, 400 MHz): δ = 2.32 (s, 3H, CH3), 3.98 (s, 6H, meta-CH3O), 4.02 (s, 3H, para-CH3O), 7.34 (s, 1H, ArCH═), 7.86 (s, 2H, Ar–H); 13C NMR (DMSO-d6, 100 MHz): δ = 11.6, 56.4, 61.2, 111.8, 117.9, 122.7, 127.7, 149.8, 152.9, 161.1, 168.5.
4-(4-(Bis(2-chloroethyl)amino)benzylidene)-3-methylisoxazol-5(4H)-one (4l)
1H NMR (300 MHz, DMSO-d6): δ = 2.21 (s, 3H, CH3), 3.81 (t, J = 6.0 Hz, 4H, 2 × CH2), 3.91 (t, J = 6.9 Hz, 4H, 2 × CH2Cl), 6.97 (d, J = 9.3 Hz, 2H, Ar–H), 7.65 (s, 1H, ArCH═), 8.45 (d, J = 8.7 Hz, 2H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 11.3, 40.9, 51.6, 110.9, 112.0, 122.1, 137.4, 150.5, 152.1, 162.1, 169.4.
(4-(4-Hydroxy-3,5-dimethoxybenzylidene)-3-methylisoxazol-5(4H)-one (4n)
1H NMR (300 MHz, DMSO-d6): δ = 2.23 (s, 3H, CH3), 3.83 (s, 6H, 2 × CH3O), 7.74 (s, 1H, ArCH═), 8.03 (s, 2H, Ar–H), 10.2 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6): δ = 11.3, 56.0, 112.7, 113.9, 123.7, 143.2, 147.5, 152.1, 162.2, 169.0.
4-((2-Hydroxynaphthalen-1-yl)methylene)-3-methylisoxazol-5(4H)-one (4q)
1H NMR (300 MHz, DMSO-d6): δ = 2.41 (s, 3H, CH3), 7.58–7.68 (m, 3H, Ar–H and ArCH═), 8.02 (d, J = 8.4 Hz, 1H, Ar–H), 8.15 (d, J = 8.4 Hz, 1H, Ar–H), 8.29 (d, J = 8.1 Hz, 1H, Ar–H), 8.45 (d, J = 7.5 Hz, 1H, Ar–H), 8.63 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6): δ = 11.3, 120.3, 124.4, 124.9, 126.6, 127.5, 128.9, 130.8, 131.4, 132.9, 148.5, 158.9, 161.9, 167.5, 169.2.
3-Methyl-4-((5-methylthiophen-2-yl)methylene)isoxazol-5(4H)-one (4s)
1H NMR (300 MHz, DMSO-d6): δ = 2.24 (s, 3H, CH3 of thiophen ring), 2.61 (s, 1H, CH3 of isoxazolone ring), 7.13 (dd, J = 0.77, 3.80 Hz, 1H, Ar–H), 8.04 (d, J = 3.8 Hz, 1H, Ar–H), 8.11 (s, 1H, ArCH═); 13C NMR (75 MHz, DMSO-d6): δ = 11.0, 15.9, 111.2, 128.3, 134.4, 141.6, 144.3, 157.1, 161.5, 168.7; IR (KBr, cm−1): ν = 3432, 1735, 1599, 1431, 1147, 1110, 1064, 998.
3-Methyl-4-((E)-3-phenylallylidene)isoxazol-5(4H)-one (4t)
1H NMR (300 MHz, DMSO-d6): δ = 2.30 (s, 1H, CH3 of isoxazolone ring), 7.46–7.66 (m, 6H, Ar–H and CH =), 7.75 (d, J = 11.7 Hz, 1H, = CH), 8.06–8.15 (m, 1H, CH═); 13C NMR (75 MHz, DMSO-d6): δ = 10.8, 116.9121.8, 128.7, 129.3, 134.8, 136.8, 149.6, 151.8, 168.7, 173.4.
3-(Chloromethyl)-4-(2-hydroxy-3-methoxybenzylidene)isoxazol-5(4H)-one (4aa)
1H NMR (300 MHz, DMSO-d6): δ = 3.85 (s, 3H, CH3O), 4.89 (s, 2H, CH2Cl), 6.89 (t, J = 8.1 Hz, 1H, Ar–H), 7.27 (d, J = 8.0 Hz, 1H, Ar–H), 8.35 (d, J = 8.2 Hz, 1H, Ar–H), 8.40 (s, 1H, ArCH═), 10.45 (s, 1H, OH); 13C NMR (75 MHz, DMSO-d6): δ = 35.3, 56.1, 113.1, 118.2, 118.7, 119.6, 123.4, 146.7, 147.7, 150.0, 161.5, 167.6; IR (KBr, cm−1): ν = 3416, 1752, 1622, 1376, 1260, 1090, 934.
3-(Chloromethyl)-4-(3,4-dimethoxybenzylidene)isoxazol-5(4H)-one (4ab)
1H NMR (300 MHz, DMSO-d6): δ = 3.84 (s, 3H, CH3O), 3.91 (s, 3H, CH3O), 4.87 (s, 2H, CH2Cl), 7.23 (d, J = 8.6 HZ, 1H, Ar–H), 8.01 (d, J = 6.7 HZ, 1H, Ar–H), 8.05 (s, 1H, Ar–H), 8.48 (s, 1H, ArCH═); 13C NMR (75 MHz, DMSO-d6): δ = 35.0, 111.4, 111.8, 125.8, 131.7, 148.4, 152.8, 155.1, 161.6, 168.3; IR (KBr, cm−1): ν = 3442, 3099, 2950, 1743, 1554, 1523, 1279, 1140, 1033, 885.
3-(Chloromethyl)-4-(3,4,5-trimethoxybenzylidene)isoxazol-5(4H)-one (4ac)
1H NMR (300 MHz, DMSO-d6): δ = 3.84 (s, 3H, CH3O), 3.86 (s, 6H, CH3O), 4.87 (s, 2H, CH2Cl), 7.97 (s, 2H, Ar–H), 8.09 (s, 1H, ArCH═); 13C NMR (75 MHz, DMSO-d6): δ = 34.9, 56.0, 112.2, 113.6, 127.6, 143.6, 152.4, 152.9, 161.6, 167.8; IR (KBr, cm−1): ν = 3434, 2941, 1725, 1570, 1426, 1349, 1258, 1125, 1112, 990.
4-(4-(Bis(2-chloroethyl)amino)benzylidene)-3-(chloromethyl)isoxazol-5(4H)-one (4ad)
1H NMR (300 MHz, DMSO-d6): δ = 3.82 (t, J = 6.7 Hz, 4H, 2 × CH2), 3.94 (t, J = 6.2 Hz, 4H, 2 × CH2Cl), 4.83 (s, 2H, CH2Cl), 7.04 (d, J = 9.2 Hz, 2H, Ar–H), 7.87 (s, 1H, ArCH═), 8.47 (d, J = 8.9 Hz, 2H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 35.2, 40.9, 51.6, 106.8, 112.4, 121.9, 137.9, 151.4, 152.9, 161.6, 169.1; IR (KBr, cm−1): ν = 3432, 1740, 1565, 1545, 1353, 1185, 1043, 933.
4-(4-(Bis(2-chloroethyl)amino)benzylidene)-3-phenylisoxazol-5(4H)-one (4al)
1H NMR (300 MHz, DMSO-d6): δ = 3.83 (t, J = 6.0 Hz, 4H, 2 × CH2), 3.95 (t, J = 6.8 Hz, 4H, 2 × CH2), 6.99 (d, J =9.1 Hz, 1H, Ar–H), 7.56 (s, 1H, ArCH═), 7.56–7.64 (m, 5H, Ar–H), 8.45 (d, J = 9.0 Hz, 2H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 40.9, 51.5, 109.3, 112.1, 121.9, 127.9, 128.7, 129.1, 130.5, 137.9, 152.6, 164.4, 169.6; IR (KBr, cm−1): ν = 3435, 1742, 1550, 1526, 1351, 1184, 1092, 828.
4-(4-hydroxybenzylidene)-3-propylisoxazol-5(4H)-one (4am)
1H NMR (500 MHz, DMSO-d6): δ = 0.99 (t, J = 7.3 Hz, 3H, CH2CH3), 1.68 (sext, J = 7.4 Hz, 2H, CH2CH3), 2.66 (t, J = 7.4, 2H, CH2), 6.95 (d, J = 8.3 Hz, 2H, Ar–H), 7.84 (s, 1H, ArCH═), 8.47 (d, J = 8.3 Hz, 2H, Ar–H); 13C NMR (125 MHz, DMSO-d6): δ = 14.1, 19.9, 27.5, 113.6, 116.6, 125.1, 138.0, 151.5, 164.3, 165.2, 169.5.
4-(4-(Dimethylamino)benzylidene)-3-propylisoxazol-5(4H)-one (4an)
1H NMR (300 MHz, DMSO-d6): δ = 0.95 (t, J = 7.4 Hz, 3H, CH3), 1.65 (sext, J = 7.4 Hz, 2H, CH2CH3), 2.55 (t, J = 7.6 HZ, 2H, CH2), 3.10 (s, 6H, N(CH3)2), 6.81 (d, J = 9.2, 2H, Ar–H), 7.58 (s, 1H, ArCH═), 8.44 (d, J = 9.0 2H, ArH); 13C NMR (75 MHz, DMSO-d6): δ = 13.7, 19.6, 27.1, 39.6, 108.3, 111.5, 120.9, 137.5, 149.9, 154.2, 164.6, 169.9; IR (KBr, cm−1): ν = 2925, 1710, 1552, 1528, 1341, 1199, 1113, 1033.
4-(3,4-Dimethoxybenzylidene)-3-propylisoxazol-5(4H)-one (4ao)
1H NMR (300 MHz, DMSO-d6): δ = 0.97 (t, J = 7.4 Hz, 3H, CH3), 1.65 (sext, J = 7.4, 2H, CH2CH3), 2.60 (t, J = 7.4 Hz, 2H, CH2), 3.81 (s, 3H, CH3O), 3.88 (s, 3H, CH3O), 7.13 (d, J = 8.6 Hz, 1H, Ar–H), 7.79 (s, 1H, ArCH═), 7.98 (dd, J = 1.6, 8.3, Hz, 1H, Ar–H), 8.45 (d, J = 1.6 Hz, 1H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 13.6, 19.2, 27.0, 55.4, 55.9, 111.4, 114.3, 115.6, 126.0, 131.0, 148.3, 151.1, 154.3, 164.6, 168.9.
3-Propyl-4-(3,4,5-trimethoxybenzylidene)isoxazol-5(4H)-one (4ap)
1H NMR (300 MHz, DMSO-d6): δ = 0.98 (t, J = 3.3 Hz, 3H, CH3), 1.68 (sext, J = 7.5 Hz, 2H, CH2CH3), 2.63 (t, J = 7.7 Hz, 2H, CH2), 3.81 (s, 3H, CH3O), 3.83 (s, 6H, 2 × CH3O), 7.87 (s, 1H, ArCH═), 7.99 (s, 2H. Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 13.6, 19.2, 27.1, 55.9, 60.4, 112.0, 116.4, 127.9, 142.9, 151.2, 152.3, 164.6, 168.5; IR (KBr, cm−1): ν = 2946, 1742, 1410, 1570, 1492, 1340, 1132, 997.
4-(4-(Bis(2-chloroethyl)amino)benzylidene)-3-propylisoxazol-5(4H)-one (4aq)
1H NMR (300 MHz, DMSO-d6): δ = 0.97 (t, J =7.4 Hz, 3H, CH3), 1.66 (sext, J = 7.4 Hz, 2H, CH2CH3), 3.82 (t, J = 6.6 Hz, 2H, 2 × CH2N), 3.92 (t, J = 6.1 Hz, 2H, 2 × CH2Cl), 6.98 (d, J = 9.2 Hz, 2H, Ar–H), 7.69 (s, 1H, ArCH═), 8.49 (d, J = 9.0 Hz, 2H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ = 13.7, 19.5, 27.0, 41.0, 51.6, 110.3, 111.5, 112.0, 122.1, 137.5, 150.1, 152.1, 164.7, 169.6. IR (KBr, cm−1): ν = 2967, 1725, 1629, 1597, 1525, 1324, 1259, 1134, 892.
Results and discussion
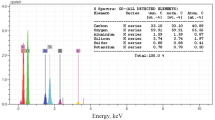
At a first step, nanosilica was synthesized according to the previously reported method [74]. Sulfuric acid immobilized on nano-SiO2 was also synthesized according to the literature [69]. To synthesize the nano-SiO2–H2SO4, a concentrated sulfuric acid was gradually added to a mortar containing nano-SiO2. The mixture was stirred for 15 min and then placed in the oven at 100 °C for 12 h. In this process, the FE-SEM images revealed that the SiO2–H2SO4 particles were formed at the nanoscale (Fig. 2). The particle size of the SiO2–H2SO4 particles typically ranges between 25 and 55 nm.

FE-SEM of synthesized nano-SiO2–H2SO4
After the synthesis of nanocatalyst, the three-component cyclocondensation (3-CC) of vanillin (1h), hydroxylamine hydrochloride (2), and ethyl acetoacetate (3a) was selected as the model reaction to establish the optimum reaction conditions (Fig. 3). Implementation of the reaction under catalyst-free conditions did not obtain pleasing results. Therefore, a catalyst was used. Satisfyingly, an expected heterocyclic product (4h) was obtained under the catalysis of nano-SiO2–H2SO4 (0.025 g) in 45% isolated yield after 30 min using water as the solvent at rt. Concomitant increase in the reaction time from 45 min to 2 h increased the reaction yield to 60%, but due to the long reaction time this change is not a desirable result. Water is close to being an ideal green solvent, preferable to alternatives such as organic solvents [1]. For this reason, water was used to carry out these 3-CCs. It should be noted that a prolonged reaction time had no significant effect on the reaction yield. Screening of the catalyst loading at rt revealed 0.05 g to be the best choice, delivering 4h in 90% isolated yields. The yield of 4h was not improved when the catalyst amount was increased from 0.05 to 0.75, 0.10, and 0.125 g at rt. The screening reaction temperatures (50, 75 °C and reflux) showed that increasing temperature did not have positive effect on the reaction yield (see red font in Fig. 3). The effects of the solvent was then investigated and found that ethanol, acetone, ethyl acetate, n-hexane, and a mixture of water–ethanol did not give better results (see blue font in Fig. 3). When the reaction was performed under solvent-free conditions, the target product was obtained in 15% isolated yield. In summary, when vanillin (1h) was treated with 2 and 3a using 0.05 g nano-SiO2–H2SO4 in water at rt for 30 min, the reaction progressed efficiently, giving 4h in 90% isolated yield (the optimized reaction conditions).

Screening of the reaction conditions of the model reaction of vanillin (1h), hydroxylamine hydrochloride (2), and ethyl acetoacetate (3a) for the synthesis of 4h in the presence of nano-SiO2–H2SO4. (Color figure online)
After the successful optimization of the reaction conditions, cyclocondensation reaction of a range of different aryl/heteroaryl aldehydes with 2 and 3a was explored, and the results are given in Table 1. Aromatic aldehydes bearing different electron-donating functional groups were reacted smoothly to form the desired α,β-unsaturated isoxazol-5(4H)-ones (4a–4p) in good to high yields (Table 1, entries 11–16). It was also found that when the heterocyclic aldehydes such as 4-oxo-4H-chromene-3-carbaldehyde, thiophene-2-carboxaldehyde, and 5-methylthiophene-2-carboxaldehyde were used to react with 2 and 3a, the target heterocyclic products (4q, 4r, and 4s) could be achieved in 86%, 90%, and 91% yields, respectively (Table 1, entries 17–19). As for steric effect, ortho-substituted benzaldehydes and 2-hydroxynaphthaldehyde were less effective than those meta-substituted or para-substituted ones, resulting in the desired products in good to high yields in 30, 10, and 40 min (Table 1, entries 6, 9, and 16), respectively. As the example of α,β-unsaturated aldehydes, cinnamaldehyde was also applied in this 3-CC and gave the 3-methyl-4-(3-phenylallylidene)isoxazol-5(4H)-one (4t) in yield of 97% (Table 1, entry 20). Nevertheless, no desired heterocyclic products were obtained for aldehydic substrates bearing electron-withdrawing group on the phenyl ring or aliphatic aldehydes. Then, instead of ethyl acetoacetate (3a), three β-ketoesters (3b–3d) as substrates were used to perform the 3-CC leading to the corresponding α,β-unsaturated isoxazol-5(4H)-ones (4u–4aq) in good to excellent yields (Table 1, entries 17–38).
The structures of the synthesized compounds were confirmed by spectral data. For example, the 1H NMR spectrum of 4l showed the doublet signals in the regions at δ 8.45 ppm (d, J = 8.7 Hz) and 6.97 (d, J = 9.3 Hz) are attributed to aromatic protons (Ar–H), and the presence of sharp singlet signal at δ 7.65 ppm is attributed to CH═ between the isoxazol and phenyl rings. The chemical shift for two chloromethylene protons (–CH2Cl–) appears as a triplet in the δ 3.91 ppm (J = 6.9 Hz), and a triplet at δ 3.81 ppm (J = 6.0 Hz) corresponds to methylene (–CH2–) linking N bound to the benzene ring. The signal of the protons of methyl group of isoxazol ring is appeared at δ 2.21 ppm as a sharp singlet signal. From the 13C NMR spectrum of 4l, the presence of signals at δ = 169.4 and 162.1 ppm corresponds to C=O and C=N of isoxazol moiety, respectively. The carbon signals of chloromethylene (–CH2Cl–) and methylene (–CH2–) groups are observed at δ 51.6 and 40.9 ppm, respectively. The carbon signal of –CH3 is observed at δ = 11.3 ppm. The other carbons exhibit peaks at their expected values (110.9, 112.0, 122.1, 137.4, 150.5, and 152.1). In comparison, the spectral data of the N-mustard compound 4ad, which is analogous to 4l, are also shown in Fig. 4.

1H and 13C NMR chemical shifts of heterocyclic compound 4ad
Furthermore, by the implementation of the reactions in water and using nano-SiO2–H2SO4 catalyst this protocol offers advantages, from the green chemistry point of view, compared to those performed in organic solvents. A literature survey on some reported approaches for the synthesis of 4-arylidene-3-methylisoxazol-5(4H)-ones is shown in Table 2. This heteroannulation reaction is preferred in some aspects, such as relatively shorter reaction times, higher efficiency, and a widespread use of β-ketoester starting materials.
Based on the previous literature [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66], the proposed mechanism for this annulation is outlined in Scheme 2. Treatment of hydroxylamine hydrochloride with β-ketoesters (3a–3d) gives the corresponding oxime derivatives C. The aldehydes activated with nano-SiO2–H2SO4 are converted to oxime arylidene intermediates (F) via the Knoevenagel reaction with oxime intermediates (D) and dehydration. The desired heterocyclic compounds (4a–4aq) were then obtained through the intramolecular O-attack cyclization and deethanolization.

Proposed reaction mechanism for the synthesis of 4-arylidene-isoxazol-5(4H)-ones (4a–4aq)
Control experiments were performed under the optimized reaction conditions to investigate insights into the reaction mechanism (Scheme 3). The first reaction (I) failed, and staining materials on TLC remained unchanged after 30 min. The subsequent reaction (II) proceeded well, and the TLC indicated that the starting materials had disappeared and a new compound had formed. The white powder was obtained and analyzed by physical data, FT-IR spectrum, and elemental analysis. It was possible to form two reaction intermediates 6 and 7. Studies show that reaction intermediate 7 was formed. In the IR spectrum, the peaks observed in regions 3458 and 1692 cm−1 indicate the formation of the oxime intermediate. The peak in the region of 3458 cm−1 confirms the hydroxyl functional group in the intermediate. Also, the ester carbonyl group appeared in the region 1692 cm−1 due to the hydrogen bond [44]. Also, elemental analysis data show that the oxime intermediate is formed, and no isoxazolone ring intermediate 6 is formed. These results showed that the oxime intermediates are involved in this 3-CC. Although the exact mechanism of the reaction unclear at this time based on the recent literature [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59] and above-mentioned control investigates, the proposed mechanism may be accepted with probability.

Control experiments to investigate the proposed reaction mechanism
The recyclability of the catalyst was investigated in the model reaction under optimized conditions (Fig. 5). After the completion of reaction, the solid product was dissolved in hot ethanol and filtered off. The catalyst remained on filter paper and heterocyclic compound in hot filtered solution. The recovered catalyst was reused three times without any significant decrease in the yield of the corresponding heterocyclic compound.

Reusability of the catalyst
Conclusions
In summary, an efficient, environmentally benign, and green approach for the synthesis of α,β-unsaturated isoxazol-5(4H)-ones from readily accessible precursors has been illustrated. The corresponding heterocyclic products were produced at room temperature in high yields and shorter reaction times. In this study, the structure of the newly synthesized α,β-unsaturated isoxazol-5(4H)-ones was confirmed on the basis of spectral data. This 3-CC approach has several additional advantages, including easy product isolation, recycling of the catalyst, minimization of waste, simplicity of operation, broad β-ketoester substrate scope, and easy work-up.
References
N. Poomathi, S. Mayakrishnan, D. Muralidharan, R. Srinivasan, P.T. Perumal, Green Chem. 17, 3362 (2015)
E. Ruijter, R. Scheffelaar, R.V.A. Orru, Angew. Chem. Int. Ed. 50, 6234 (2011)
H. Kiyani, F. Ghorbani, J. Saudi Chem. Soc. 18, 689 (2014)
S.P.N. Sudhan, R. Nasir Ahmed, H. Kiyani, S. Sheik Mansoor, J. Saudi Chem. Soc. 22, 269 (2018)
H. Kiyani, Curr. Org. Synth. 15, 1043 (2018)
Y. Gu, W. Huang, S. Chen, X. Wang, Org. Lett. 14, 4285 (2018)
J. Xu, W. Huang, R. Bai, Y. Queneau, F. Jérôme, Y. Gu, Green Chem. 21, 2061 (2019)
N. Agrawal, P. Mishra, Med. Chem. Res. 27, 1309 (2018)
S.M. Gomha, M.G. Badrey, M.M. Abdalla, R.K. Arafa, Med. Chem. Commun. 5, 1685 (2014)
S.S. Wazalwar, A.R. Banpurkar, F. Perdih, J. Mol. Struct. 1150, 258 (2017)
A.R. Banpurkar, S.S. Wazalwar, F. Perdih, Bull. Chem. Soc. Ethiop. 32, 249 (2018)
V.S. Konkala, P.K. Dubey, J. Heterocycl. Chem. 54, 2483 (2017)
S.J. Kim, J. Yang, S. Lee, C. Park, D. Kang, J. Akter, S. Ullah, Y.J. Kim, P. Chun, H.R. Moon, Bioorg. Med. Chem. 26, 3882 (2018)
C. Bustos, E. Molins, J.G. Carcamo, M.N. Aguilar, C. Sanchez, I. Moreno-Villoslada, H. Nishide, X. Zarate, E. Schott, New J. Chem. 40, 2156 (2016)
M. Lavanya, M. Jagadeesh, J. Hari Babu, R. Karvembu, H.K. Rashmi, P. Uma Maheswari Devi, A.V. Reddy, Inorganica Chimica Acta 469, 76 (2018)
N. Panathur, N. Gokhale, U. Dalimba, P.V. Koushik, P. Yogeeswari, D. Sriram, Bioorg. Med. Chem. Lett. 25, 2768 (2015)
B. Kafle, N.G. Aher, D. Khadka, H. Park, H. Cho, Chem. Asian J. 6, 2073 (2011)
B. Kafle, H. Cho, Bull. Korean Chem. Soc. 33, 275 (2012)
Y. Kazui, S. Fujii, A. Yamada, M. Ishigami-Yuasa, H. Kagechik, A. Tanatani, Bioorg. Med. Chem. 26, 5118 (2018)
A. Ishioka, K. Tanatani, Y.Hashimoto Nagasawa, Bioorg. Med. Chem. Lett. 13, 2655 (2003)
M. Tang, S.I. Odejinmi, Y.M. Allette, H. Vankayalapati, K. Lai, Bioorg. Med. Chem. 19, 5886 (2011)
S. Breuer, M.W. Chang, J. Yuan, B.E. Torbett, J. Med. Chem. 55, 4968 (2012)
Ş.G. Kömürcü, S. Rollas, N. Yilmaz, A. Çevikbaş, Drug Metabol. Drug Interact. 12, 161 (1995)
W. Hallenbach, O. Guth, T. Seitz, H.J. Wrolowsky, P. Desbordes, U. Wachendorff-Neumann, P. Dahmen, E. Voerste, P. Lösel, O. Malssm, R. Rama, H. Hadano, US Patent, Pub. No.: US 2012/0065063A1 (2012)
W. Hallenbach, O. Guth, T. Seitz, H.J. Wrolowsky, P. Desbordes, U. Wachendorff-Neumann, P. Dahmen, E. Voerste, P. Lösel, O. Malssm, R. Rama, H. Hadano, WIPO Patent Application WO/2011/161035A1 (2011)
A.H. Reshak, S. Azam, Mater. Sci. Semicond. Process. 30, 197 (2015)
X. Zhang, X. Jiang, Y. Li, Z. Lin, G. Zhang, Y. Wu, CrystEngComm 17, 7316 (2015)
D. Jiang, Z. Xue, Y. Li, H. Liu, W. Yang, J. Mater. Chem. C 1, 5694 (2013)
A.F. da Silva, A.A.G. Fernandes, S. Thurow, M.L. Stivanin, I.D. Jurberg, Synthesis 50, 2473 (2018)
X.H. Zhang, L.Y. Wang, Y.H. Zhan, Y.L. Fu, G.H. Zhai, Z.Y. Wen, J. Mol. Struct. 994, 371 (2011)
X.H. Zhang, Y.H. Zhan, D. Chen, F. Wang, L.Y. Wang, Dyes Pigments 93, 1408 (2012)
E. Aret, H. Meekes, E. Vlieg, G. Deroover, Dyes Pigments 72, 339 (2007)
T. Ghosh, A. Gopal, A. Saeki, S. Sekic, V.C. Nair, Phys. Chem. Chem. Phys. 17, 10630 (2015)
F. Saikh, J. Das, S. Ghosh, Terahedron Lett. 54, 4679 (2013)
H. Kiyani, F. Ghorbani, J. Saudi Chem. Soc. 21, S112 (2017)
H. Kiyani, F. Ghorbani, Res. Chem. Intermed. 41, 7847 (2015)
H. Kiyani, M. Jabbari, A. Mosallanezhad, Jordan J. Chem. 9, 279 (2014)
H. Kiyani, H. Darbandi, A. Mosallanezhad, F. Ghorbani, Res. Chem. Intermed. 41, 7561 (2015)
H. Kiyani, F. Ghorbani, A. Kanaani, D. Ajloo, M. Vakili, Res. Chem. Intermed. 41, 7739 (2015)
S.N. Maddila, S. Maddila, W.E. van Zyl, S.B. Jonnalagadda, Res. Chem. Intermed. 42, 2553 (2016)
R.H. Vekariya, K.D. Patel, H.D. Patel, Res. Chem. Intermed. 42, 7559 (2016)
J. Safari, M. Ahmadzadeh, Z. Zarnegar, Catal. Commun. 86, 91 (2016)
Q. Liu, Y.N. Zhang, Bull. Korean Chem. Soc. 32, 3559 (2011)
H. Kiyani, F. Ghorbani, Res. Chem. Intermed. 41, 2653 (2015)
M. Ahmadzadeh, Z. Zarnegar, J. Safari, Green Chem. Lett. Rev. 11, 78 (2018)
H. Kiyani, H.A. Samimi, Chiang Mai J. Sci. 44, 1011 (2017)
A.B. Rikani, D. Setamdideh, Orient. J. Chem. 32, 1433 (2016)
R.H. Vekariya, H.D. Patel, Indian J. Chem. 56B, 890 (2017)
M.S. Patil, C. Mudalian, G.U. Chaturbhuj, Tetrahedron Lett. 58, 3256 (2017)
R. Laroum, A. Debache, Synth. Commun. 48, 1876 (2018)
T. Lohar, A. Kumbhar, M. Barge, R. Salunkhe, J. Mol. Liq. 224, 1102 (2016)
Q. Liu, X. Hou, Phosphorus Sulfur Silicon Relat. Elem. 187, 448 (2012)
H. Kiyani, F. Ghorbani, Res. Chem. Intermed. 42, 6831 (2016)
K. Ablajan, H. Xiamuxi, Synth. Commun. 42, 1128 (2012)
K. Ablajan, H. Xiamuxi, Chin. Chem. Lett. 22, 151 (2011)
Q.F. Cheng, X.Y. Liu, Q.F. Wang, L.S. Liu, W.J. Liu, Q. Lin, X.J. Yang, Chin. J. Org. Chem. 29, 1267 (2009)
H. Kiyani, A. Mosallanezhad, Curr. Org. Synth. 15, 715 (2018)
A. Mosallanezhad, H. Kiyani, Orbital Electron J. Chem. 10, 133 (2018)
Q. Liu, R.T. Wu, J. Chem. Res. 35, 598 (2011)
A. Ahad, M. Farooqui, Int. J. Chem Tech Res. 10, 269 (2017)
G.T. Pawar, S.P. Gadekar, B.R. Arbad, M.K. Lande, Bull. Chem. React. Eng. Catal. 12, 32 (2017)
S.P. Vaidya, G. Shridhar, S. Ladage, L. Ravishankar, Curr. Green Chem. 3, 160 (2016)
M.G. Dekamin, S.Z. Peyman, Monatsh. Chem. 147, 445 (2016)
S.A. Pourmousavi, H.R. Fattahi, F. Ghorbani, A. Kanaani, D. Ajloo, J. Iran. Chem. Soc. 15, 455 (2018)
D. Setamdideh, J. Serb. Chem. Soc. 81, 971 (2016)
G. Ferouani, A. Nacer, N. Ameur, R. Bachir, C. Ziani-Cherif, J. Chin. Chem. Soc. 65, 459 (2018)
A. Mosallanezhad, H. Kiyani, Curr. Organocatal. 6, 28 (2019)
S. Rasheed, D.N. Rao, A.S. Reddy, R. Shankar, P. Das, Rsc Adv. 5, 10567 (2015)
V.K. Rajput, B. Mukhopadhyay, Tetrahedron Lett. 47, 5939 (2006)
J. Zhang, B. Zhang, J. Zhou, J. Li, C. Shi, T. Huang, Z. Wang, J. Tang, J. Carbohydr. Chem. 30, 165 (2011)
J. Zhang, B. Zhang, J. Zhou, H. Chen, J. Li, G. Yang, Z. Wang, J. Tang, J. Carbohydr. Chem. 32, 380 (2013)
J.F. Zhou, X. Chen, Q.B. Wang, B. Zhang, L.Y. Zhang, A. Yusulf, Z.F. Wang, J.B. Zhanga, J. Tang, Chin. Chem. Lett. 21, 922 (2010)
O. Rosati, F. Messin, A. Pelosi, M. Curini, V. Petrucci, J. Gertsch, A. Chicca, Eur. J. Med. Chem. 85, 77 (2014)
K.S. Rao, K. El-Hami, T. Kodaki, K. Matsushige, K. Makino, J. Colloid Interface Sci. 289, 125 (2005)
Acknowledgements
The authors are thankful to Damghan University Research Council for financial support of this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghorbani, F., Kiyani, H. & Pourmousavi, S.A. Facile and expedient synthesis of α,β-unsaturated isoxazol-5(4H)-ones under mild conditions. Res Chem Intermed 46, 943–959 (2020). https://doi.org/10.1007/s11164-019-03999-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-019-03999-7