
Abstracts
Most radiation from the human body belongs to the long-wavelength infrared region (mainly at wavelengths of 10–12 μm), which allows infrared (IR) sensors to distinguish humans or animals in the dark. One of the important components of such IR sensors is the IR window, which requires high transparency in the infrared region. ZnS is a most desirable substance for such windows due to its high transparency and high thermal stability. Herein, we report a low-cost environmentally friendly synthesis method for ZnS powder and its sintering for use as window material. Using only elemental precursors, Zn and S, submicrometer-sized ZnS powder with high phase purity was obtained. Spark plasma sintering was applied to compact the ZnS powder, and the resulting ceramic was analyzed by scanning electron microscopy, Fourier-transform infrared spectroscopy, Raman spectroscopy, and X-ray diffraction. We also examined the effect of an additive during sintering and found that LiF functions well as a sintering aid for the ZnS compact.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most radiation from the human body belongs to the mid- and long-wavelength infrared (IR) region (mainly at wavelengths of 5–20 μm), which allows IR sensors to distinguish humans or animals in the dark. An IR sensor is composed of an IR detector plus optical components such as windows and lenses. The latter accounts for more than 30% of the whole device cost, so reducing this cost is of importance in the related industry. Materials available for optical components include ZnS, ZnSe, Ge, sapphire, etc. Among them, ZnS (chalcogenide material) is most desirable due to its high transparency in the 3–12 μm wavelength range and high thermal stability up to ~ 600 °C [1].
A number of synthetic methods have been reported for ZnS materials, for example, hydrothermal synthesis [2], precipitation process [3, 4], solid-state synthesis [5], vapor-phase synthesis including spray pyrolysis, and chemical vapor synthesis [6, 7]. Mechanochemical synthesis, however, has been exploited for ZnS in only limited cases [8,9,10], although it has been widely used for other chalcogenide materials [11]. Mechanochemical synthesis, especially with elemental precursors in dry (solvent-free) condition, offers advantages over other methods, such as cost efficiency, environmental friendliness, and high throughput [12].
Fabrication of transparent ceramic windows has been carried out by a variety of sintering methods that include pressureless sintering (PS), hot pressing (HP), hot isostatic pressing (HIP), hot uniaxial pressing (HUP), and spark plasma sintering (SPS) [13,14,15]. To form a highly transparent window, it is important to control the phase transformation, birefringence, grain growth, and formation of defects (including pores and impurities). Therefore, an appropriate process needs to be chosen for each specific material, and sometimes use of additives can enhance the sintering process [16]. Recent studies showed that spark plasma sintering (SPS) is a powerful method to obtain transparent ceramics [17], offering advantages over other sintering methods in terms of relatively low sintering temperature and short reaction time.
We report herein synthesis of ZnS powder by a mechanochemical method using elemental precursors, Zn and S, in dry condition. This process not only avoids formation of byproducts but also reduces consumption of time and materials caused by workup steps such as washing, centrifugation, and drying. To obtain ZnS materials with quality suitable for use as IR windows, the crystal structure and elemental purity were carefully monitored while optimization the milling conditions (e.g., types of milling jar and balls, milling velocity and time, and ball-to-powder ratio). Sintering of the ZnS powders was achieved by SPS technique. The effect of additives, viz. LiF and CaF2, on the sintered body was also examined.
Experimental
Synthesis of ZnS powder by mechanochemical method
Mechanochemical synthesis was conducted using a planetary ball-mill machine (Fritsch GmbH, Pulverisette 6 classic line) using elemental precursors, zinc (Alfa Aesar, 99.9%, ~ 100 mesh) and sulfur (Sigma Aldrich, 99.99%). A stoichiometric mixture of Zn and S (3–10 g in total) was charged into a round-ended milling jar (80 mL volume) in a glovebox filled with argon, then milling balls were added. The ball-to-powder (charge) ratio was fixed at 16.7:1 by weight. The milling environment was controlled inert, since the ZnS surface can be oxidized by air to form ZnSO4 under high-energy milling conditions [10]. Planetary ball-milling was carried out in solvent-free condition at speed of 500 rpm for 14–16 h, with 1 h of continuous milling and 10 min interval; aliquots (~ 0.1 g) were taken intermittently for evaluation. As additives, LiF (98.0+%, Wako) and CaF2 (99.95%, CERAC) were purchased and used as received, being added to as-synthesized ZnS powder at 2 wt%, followed by milling of the mixture under the same condition for 1 h. All powders were heat-treated in a tube furnace at 500 °C for 1 h in sulfur-rich atmosphere (Ar with 5% H2S) prior to sintering.
Fabrication of ZnS windows by SPS technique
Spark plasma sintering was carried out using a HP-D5 (FCT System GmbH). Graphite was used as a mold and punch (20 mm diameter) material during SPS. ZnS powder (~ 1.8 g) was placed in between boron nitride powder (2 g, 98.0%, Sigma-Aldrich) and sintered to minimize carbon contamination from the graphite mold following literature procedure [18]. The sample was heated from room temperature to final temperature (920 °C) at rate of 100 °C/min, then held at the final temperature under load of 50 MPa for 10 min, followed by water cooling. The boron nitride powder was removed from the ZnS sintered body (~ 20 mm diameter, ~ 2 mm thickness), and the sintered ZnS was polished to form an IR-transparent disc (average thickness ~ 0.4 mm).
Characterization
Morphological observation of powders and sintered bodies was conducted by field-emission scanning electron microscopy (FE-SEM; Inspect F50, FEI Company). Crystal structure was studied by X-ray diffraction (XRD) analysis (D8 Advance, Bruker Corporation) with Cu Kα radiation (λ = 0.154178 nm), and peaks were calibrated with respective to a Si standard sample (SRM 640d). Particle size was obtained using a particle size analyzer (Microtrac, Nanotrac Wave) from powder suspension in water. Elemental analysis was conducted by X-ray fluorescence (XRF, RIX2100, Bruker Corporation). Compact density was determined by applying Archimedes’ principle with immersion in water at 22 °C (Shimadzu AX200/401 SMK, accuracy ±0.001 g). The compacity was calculated based on the density of würtzite phase (4.090 g/cm3). Fourier-transform infrared (FT-IR) spectroscopy was carried out at room temperature using a VERTEX 80v FT-IR spectrometer in the range of 2–16 μm. Raman spectra were obtained using an inVia (Renishaw) Raman microscope equipped with a confocal microscope. Samples were excited at room temperature using a 532-nm neodymium-doped yttrium aluminum garnet (Nd:YAG) laser or 785-nm diode laser.
Results and discussion
To obtain ZnS powders with high purity, proper selection of milling jar and ball materials is required. Among available milling materials, we selected three different ones, viz. sintered corundum, stainless steel, and zirconium oxide, which have fairly good resistance to abrasion. For the stainless-steel jar, balls made of the same material were used, while for the sintered corundum and zirconium oxide jars, balls made of zirconium oxide were used. The ball-to-powder ratio was varied from 25:1 to 5:1 (by weight) and found to be optimal at 16.7:1. Therefore, the ball-to-powder ratio was fixed at 16.7:1 during further optimization. For comparison of the different milling materials, planetary ball-milling was carried out under the exactly same condition for all three materials (milling speed 500 rpm, reaction time 14 h with 60-min runs and 10-min breaks). Figure 1a shows the XRD patterns of products obtained using different milling materials. Using all three, cubic ZnS was found as major phase with minimal or no trace of elemental Zn and S. Major peaks were observed at 2θ values of 28.51°, 47.61°, and 56.41°, corresponding to diffraction from (1 1 1), (2 2 0), and (3 1 1) planes of cubic phase ZnS according to Joint Committee on Powder Diffraction Standards (JCPDS) card no. 05-0566. Minor peaks belonging to hexagonal phase (JCPDS card no. 36-1450) were detected for all three samples.

a X-ray diffraction patterns and b particle size distribution and SEM image (scale bar 1 μm) of ZnS obtained using different milling materials: (i) sintered corundum, (ii) stainless steel, and (iii) zirconium oxide
To compare the particle size of ZnS obtained using the different milling jar materials (sintered corundum, stainless steel, and zirconium oxide), particle size analysis was performed on each product. Figure 1b shows the size distribution results with inset SEM images. Although all samples showed agglomerates of several micrometers (1–6 μm), both the size analysis and SEM images suggested that most particles were of submicrometer size. Among the different jar materials, the zirconium oxide jar appeared to provide the finest particles with average size of 300 nm.
To compare the abrasion resistance of the different milling materials, elemental analysis was conducted for the three different products. Table 1 presents the molar ratio (in %) of the major components of each product along with impurities, which were generated from the milling material by abrasion and therefore differed for the different milling jars. The product obtained using the zirconium oxide jar was found to contain the lowest amount of impurity (Zr, ~ 0.025%), whereas that obtained using the sintered corundum jar had the highest amount of impurity (Al, ~ 3.56%). Previously, Balaz et al. reported synthesis of ZnS by ball milling with hardened-steel materials and observed that the product was contaminated by up to 0.14% iron after 90 min of milling at ~ 700 rpm [10]. We also found that the stainless-steel jar was abraded and contaminated the resulting product at about 0.063% under our milling condition. Therefore, we concluded that jars and balls made of zirconium oxide were most suitable for mechanochemical synthesis of ZnS powders and used them for further optimization.
Using the zirconium oxide jar and balls, we milled for more than 14 h to obtain ZnS powder with high purity and ultrafine size (below hundreds of nanometers). Figure 2a shows X-ray diffraction patterns of products obtained after ball milling for 14, 15, and 16 h at 500 rpm, clearly revealing that ZnS was completely formed after 15 h with no trace of residual precursor elements, Zn and S. It was also found that cubic phase of ZnS was preferentially formed (by phase fitting of XRD peaks with respect to that of Si). Particle grinding occurred for 16 h of milling as a result of the prolonged milling time, as evidenced by broadening of XRD peaks. The crystalline size of each product was calculated using the Debye–Scherrer equation [19] using the full-width at half-maximum (FWHM) of the (1 1 1) plane [FWHM = 0.642° (14 h), 0.681° (15 h), and 0.889° (16 h)]. The crystalline size of ZnS obtained after 16 h of milling was reduced to 9.2 nm from 12.1 and 12.8 nm after 14 and 15 h of milling, respectively. Furthermore, the phase purity of the obtained product was very high, since only a very small amount of hexagonal phase was found in the XRD patterns.

a X-ray diffraction patterns and b particle size distribution and SEM image (scale bar 1 μm) of ZnS after milling for (i) 14 h, (ii) 15 h, and (iii) 16 h at 500 rpm
To further prove the particle size reduction for extended milling time, we performed particle size analysis on the products obtained after 14, 15, and 16 h of milling. Figure 2b shows the particle size distribution and FE-SEM images of ZnS milled for 14, 15, and 16 h. The ZnS product obtained after 14 h showed nano- to micrometer-sized particles, whereas that obtained after 16 h showed very fine particle size with average of 196 nm. The ZnS powder obtained by the optimized method will be highly suitable for fabrication of IR windows due to its fine size and good phase and elemental purity. Therefore, we carried out sintering on ZnS powder obtained after 16 h of milling.
Spark plasma sintering (SPS, also known as pulsed electric current sintering) was applied to the ZnS powder to form IR-transparent ceramics. During SPS, ZnS powder was packed into a graphite mold and pulsed DC current directly passed through the graphite mold while uniaxial pressure was applied by a plunger. Since the heat generation during SPS is internal, SPS offers a very high heating rate and hence the sintering process can be very fast (typically within 10 min). Therefore, this technique is particularly useful for sintering fine powders, since short sintering time can minimize the grain growth of fine powders often observed during conventional hot pressing. However, when we applied SPS to our ZnS powder, severe carbon contamination occurred, preventing FT-IR transmittance measurements.
Such carbon contamination during SPS is not unusual, hence several techniques are available to reduce it, including pressureless sintering of powder prior to SPS and use of a physical carbon diffusion barrier [20]. Among barrier materials for graphite, boron nitride has been proven to be very effective for blocking carbon diffusion during SPS [18]. It has high electrical resistivity at elevated temperature [21] as well as good thermal conductivity and thermal shock resistance. Therefore, a boron nitride layer was placed on the top and bottom of our ZnS powder compact to avoid its direct contact with the graphite mold.
To examine the effect of sintering aids (additives) during SPS treatment, two additives, LiF and CaF2, were chosen based on a literature search for sintering aids [22,23,24,25]. Each additive (2 wt%) was mixed with ZnS powder and dry-milled at 500 rpm for 1 h. Under the same sintering condition (920 °C for 10 min with load of 50 MPa), three disc samples were prepared: additive-free (ZnS), with CaF2 (ZnS–CaF2), and with LiF (ZnS–LiF).
The microstructure of each sintered body was examined by FE-SEM (Fig. 3). The first row of Fig. 3 shows fracture images, while the second row shows top views of ZnS, Zn–CaF2, and ZnS–LiF disc samples after polishing. It was observed that the ZnS disc had inhomogeneous microstructure with a large number of internal pores (of less than 1 μm) distributed throughout. This poor microstructure is surmised to arise from insufficient sintering of the starting powder. On the other hand, use of an additive (LiF or CaF2) not only reduced the pore volume but also enhanced the sintering, indicating that both additives functioned well as flux during ZnS sintering. When ZnS–LiF and ZnS–CaF2 were compared, grain sizes were larger for ZnS–CaF2 but the size distribution was slightly narrower for ZnS–LiF. The fracture image of ZnS–CaF2 (Fig. 3b) also showed presence of submicrometer-size grains (marked with red arrows in the figure), which may imply low solubility of CaF2 in ZnS (as the small grains might comprise undissolved CaF2 particles). It is plausible that the sintering condition was insufficient to dissolve the CaF2 additive, since its melting point is 1418 °C at ambient pressure (0.1 MPa). Nevertheless, ZnS–CaF2 showed less voids compared with ZnS, indicating that sintering could be accelerated by much smaller amounts of CaF2 than 2 wt%.

FE-SEM images: fracture views (first row) and top views (second row) of a, d ZnS, b, e ZnS–CaF2, and c, f ZnS–LiF samples (scale bar 10 μm)
The density of three different disc samples was obtained by the Archimedes method [26], and the results are provided in Table 2. In good agreement with the microstructure observations, ZnS–LiF showed the highest density (99.8%), followed by ZnS–CaF2 (98.9%), while ZnS showed the lowest density (97.1%). Based on the SEM observations and density measurements, we conclude that, among the three samples, ZnS–LiF had the least pore volume and highest density and is likely most suitable for use in IR-transparent windows.
Figure 4 presents the FT-IR transmittance (in the range of 2–16 μm) of all three samples. As expected, ZnS–LiF exhibited the highest transmittance, whereas ZnS showed the lowest. ZnS had particularly low transmittance in the mid-wavelength infrared (MW-IR) range (3–8 μm), which may originate from scattering by internal pores and grain boundaries. In the long-wavelength infrared (LW-IR) range (8–15 μm), ZnS–LiF exhibited the highest transmittance, followed by ZnS–CaF2 and then ZnS. Notably, strong absorption peaks were found at ~ 9 and ~ 13.5 μm only for ZnS–LiF and ZnS–CaF2 samples. The peak at ~ 9 μm appears to be associated with carbon contamination (C–C, C–O, or C–S bonds), as other researchers also reported the same peak for samples sintered in a graphite mold [14, 19]. The peak at ~ 13.5 μm is not well understood, but the fact that it was found only for samples with additives led us to consider it to originate from carbonation of fluoride compounds (e.g., C–F, O–C–F, etc.). Earlier we discussed use of boron nitride as a barrier to carbon diffusion [18]. Although data are not shown, when the boron nitride layer was not used during SPS, the carbon contamination was much more severe, so that we could not observe any infrared transmittance for any sample. Use of boron nitride afforded higher transmittance for all samples and no carbon contamination for the undoped sample, ZnS (see the flat line at ~ 9 μm in Fig. 4a).

Infrared transmittance obtained for a ZnS, b ZnS–CaF2, and c ZnS–LiF discs (intensity normalized by thickness of each disc)
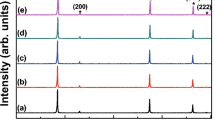
To further understand the chemical and phase properties of the sintered samples, we carried out Raman spectroscopy and XRD analysis. Figure 5a presents the Raman spectra of ZnS, ZnS–CaF2, and ZnS–LiF (bottom to top). Two distinctive peaks at ~ 350 and ~ 670 cm−1 and a broad peak at 350–450 cm−1 are seen for all samples. The first two peaks can be identified as due to first-order longitudinal optical (1LO) and second-order LO (2LO) phonon scattering of ZnS [27]. The broad peak was also seen by other researchers and has been identified as a two-phonon density of states of ZnS [28]. A small peak at ~ 640 cm−1 was also found (most distinctively with ZnS), which may be associated with defects of ZnS [27]. Presence of ZnO was confirmed by Raman spectroscopy, since its scattering peaks at 575 and 1150 cm−1 are quite sensitive [29]. Only a tiny peak at 575 cm−1 was found with ZnS–LiF, presumably indicating partial oxidation.

a Raman spectroscopy and b X-ray diffraction patterns of (i) ZnS, (ii) ZnS–CaF2, and (iii) ZnS–LiF discs
Figure 5b shows the XRD patterns of the three samples. Albeit to different degrees, phase transformation from cubic (zincblende) to hexagonal (würtzite) phase occurred for all samples. This phase transformation is not desirable, since the hexagonal phase is birefringent (optically anisotropic) whereas the cubic phase is not [30]. Formation of hexagonal phase was not expected because this phase transformation is known to occur at 1020 °C [31], much higher than our applied temperature of 920 °C. We assume that the heat generated by the pulsed DC current during SPS was not uniform, so that the local temperature inside the ceramic may have exceeded the set temperature. Nevertheless, the relative ratio of hexagonal to cubic phase lay in the order ZnS–CaF2 < ZnS–LiF < ZnS (complete hexagonal phase), suggesting that the additives, especially CaF2, acted as inhibitors of the phase transformation, representing another advantage of their use for production of transparent ZnS ceramics.
Conclusions
We report mechanochemical synthesis of ZnS powder and its use for fabrication of transparent ceramics. The synthesis of ZnS powder was optimized in terms of minimal contamination by milling materials and the particle size distribution. A jar and balls made of zirconium oxide and milling condition of 16 h at speed of 500 rpm were found to be most suitable to obtain ZnS powder with high purity and ultrafine size (~ 200 nm). Another feature of this method is its environmental friendliness, since it includes only the elemental precursors, Zn and S, in the absence of any solvent. Spark plasma sintering was applied to the powder compact to afford transparent ceramic. During SPS, additives such as LiF and CaF2 were included in the ZnS powder, being proven to aid sintering and partially inhibit the (cubic to hexagonal) phase transformation. Among the additives, LiF was found to be preferable, providing the highest compacity of 99.8% and maximum transmittance of ~ 55%. The temperature applied during SPS appeared to be slightly higher than the set value, as determined by the high degree of phase transformation and large grain sizes, so we believe that the optical quality of such transparent ceramics could be further improved by optimizing the sintering condition.
References
D.C. Harris, Infrared Phys. Technol. 39, 185 (1998)
L. Yin, D. Wang, J. Huang, L. Cao, H. Ouyang, X. Yong, J. Alloys Compd. 664, 476 (2016)
A. Celikkaya, M. Akinc, J. Am. Ceram. Soc. 73, 245 (1990)
Y.-P. Zhu, J. Li, T.-Y. Ma, Y.-P. Liu, G. Du, Z.-Y. Yuan, J. Mater. Chem. A 2, 1093 (2014)
F. Chen, Y. Cao, D. Jia, Ceram. Int. 41, 6645 (2015)
S. Liu, H. Zhang, M.T. Swihart, Nanotechnology 20, 235603 (2009)
X. Fang, T. Zhai, U.K. Gautam, L. Li, L. Wu, Y. Bando, D. Golberg, Prog. Mater. Sci. 56, 175 (2011)
C.S. Pathak, D.D. Mishra, V. Agarawala, M.K. Mandal, Ind. J. Phys. 86, 777 (2012)
C.S. Pathak, V. Agarwala, M.K. Mandal, Phys. B Condens. Matter 407, 3309 (2012)
P. Balaz, M. Balintova, Z. Bastl, J. Briancin, V. Sepelak, Solid State Ionics 101, 45 (1997)
P.G. McCormick, T. Tsuzuki, J.S. Robinson, J. Ding, Adv. Mater. 13, 1008 (2001)
B.I. Park, Y. Hwang, S.Y. Lee, J.S. Lee, J.K. Park, J. Jeong, J.Y. Kim, B. Kim, S.H. Cho, D.K. Lee, Nanoscale 6, 11703 (2014)
P. Biswas, R.S. Kumar, P. Ramavath, V. Mahendar, G.V.N. Rao, U.S. Hareesh, R. Johnson, J. Alloys Compd. 496, 273 (2010)
C. Chlique, G. Delaizir, O. Merdrignac-Conanec, C. Roucau, M. Dolle, P. Rozier, V. Bouquet, X.H. Zhang, Opt. Mater. 33, 706 (2011)
Y.Z. Chen, L. Zhang, J. Zhang, P. Liu, T.Y. Zhou, H.X. Zhang, D.M. Gong, D.Y. Tang, D.Y. Shen, Opt. Mater. 50, 36 (2015)
N. Frage, S. Kalabukhov, N. Sverdlov, V. Kasiyan, A. Rothman, M.P. Dariel, Ceram. Int. 38, 5513 (2012)
Z.A. Munir, U. Anselmi-Tamburini, M. Ohyanagi, J. Mater. Sci. 41, 763 (2006)
D. Giuntini, J. Raethel, M. Herrmann, A. Michaelis, E.A. Olevsky, J. Am. Ceram. Soc. 98, 3529 (2015)
C. Chlique, O. Merdrignac-Conanec, N. Hakmeh, X. Zhang, J.-L. Adam, S. Bhandarkar, J. Am. Ceram. Soc. 96, 3070 (2013)
A. Bertrand, J. Carreaud, G. Delaizir, J.R. Duclere, M. Colas, J. Cornette, M. Vandenhende, V. Couderc, P. Thomas, J. Am. Ceram. Soc. 97, 163 (2014)
L.G. Carpenter, P.J. Kirby, J. Phys. D Appl. Phys. 15, 1143 (1982)
R. Marder, R. Chaim, G. Chevallier, C. Estournès, J. Eur. Ceram. Soc. 31, 1057 (2011)
W.S. Xia, L.X. Li, P. Zhang, P.F. Ning, Mater. Lett. 65, 3317 (2011)
M. Pollet, S. Marinel, J. Eur. Ceram. Soc. 23, 1925 (2003)
Y. Xiong, Z.Y. Fu, H. Wang, Y.C. Wang, Q.J. Zhang, Mater. Sci. Eng. B 123, 57 (2005)
G.G. Moraes, I.E.F. Pozzobom, C.P. Fernandes, A.P.N. de Oliveira, Chem. Eng. Trans. 43, 1801 (2015)
Y.Y. Luo, G.T. Duan, G.H. Li, Appl. Phys. Lett. 90, 201911 (2007)
Z.G. Chen, L.N. Cheng, J. Zou, Cryst. Eng. Commun. 13, 5885 (2011)
X.M. Shuai, W.Z. Shen, J. Phys. Chem. C 115, 6415 (2011)
S.B. Qadri, E.F. Skelton, D. Hsu, A.D. Dinsmore, J. Yang, H.F. Gray, B.R. Ratna, Phys. Rev. B 60, 9191 (1999)
A.K. Kole, P. Kumbhakar, Results Phys. 2, 150 (2012)
Acknowledgements
This work was supported by institutional funding from the Korea Institute of Science and Technology (Grant No. 2E26960), the KIST-UNIST partnership program (1.160097.01/2V05150), and the R&D Convergence Program of NST (National Research Council of Science & Technology) of Republic of Korea (Grant No. 2N42860).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Ahn, HY., Choi, W.J., Lee, S.Y. et al. Mechanochemical synthesis of ZnS for fabrication of transparent ceramics. Res Chem Intermed 44, 4721–4731 (2018). https://doi.org/10.1007/s11164-018-3277-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-018-3277-4