
Abstract
A simple and efficient procedure for synthesis of 2-substituted benzimidazole and benzothiazole has been developed by using sulfonic-acid-functionalized activated carbon as heterogeneous catalyst. The activated material was prepared from matured tea leaf in presence of phosphoric acid as activating agent. The final catalyst was prepared by anchoring –SO3H group on the surface of the activated carbon. The catalyst could be easily recovered and reused for more than three catalytic cycles without significant loss in catalytic activity. The catalytic performance of the catalyst was found to be superior to that of a similar catalyst prepared from montmorillonite K10.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Benzimidazoles and benzothiazoles are considered to be attractive building blocks by synthetic chemists due to their presence as active substructures in biologically significant compounds. These structural motifs are associated with a wide variety of pharmacological activities such as antiviral, antifungal, anticancer, antiulcer, antihypertensive, and antihistaminic effects [1,2,3,4,5,6,7,8]. Benzimidazole derivatives exhibit considerable activity against several viruses such as human immunodeficiency virus (HIV) and herpes simplex virus type 1 (HSV-1), and also act as antitumor and antimicrobial agents [9,10,11,12]. Benzothiazole derivatives have applications as enzyme inhibitors, antitumor agents, fluorescent materials, and in vivo imaging agents [13,14,15,16].
Due to the extensive utility of these heterocycles, a variety of methods for their synthesis have been widely reported. The most common synthetic method involves condensation of 1,2-phenylenediamine or 2-aminothiophenol with aldehydes [17,18,19]. In case of some other synthetic approaches, orthoesters [20, 21], carboxylic acid [22,23,24], alcohols [25], acid chlorides [26] or diketones [27, 28] have also been used instead of aldehydes.
Recently, Martins et al. [29] reported a procedure for synthesis of benzimidazole using Ce(NO3)3·6H2O as promoter in dimethylformamide (DMF). Shirini et al. [30] and coworkers synthesized benzimidazole derivatives using 1,1′-disulfo-[2,2′-bipyridine]-1,1′-diium chloride ionic liquid as homogeneous catalyst. Hanoon et al. [31] described synthesis of benzimidazole derivatives in presence of ionic-liquid-functionalized reduced graphene oxide as catalyst. Another method using graphene oxide as catalyst for synthesis of benzimidazoles and benzothiazoles was reported by Dhopte [32]. Use of CuI nanoparticles for synthesis of 2-substituted benzimidazoles requires use of molecular oxygen as oxidant [33]. Although there are several reports on synthesis of these two heterocycles, most of the traditional synthetic methods suffer from one or more drawbacks, such as high temperature, long reaction time, low yield of product, use of toxic organic solvents, harsh reaction condition, use of expensive and hazardous catalyst, etc. Taking into account the various drawbacks of existing synthetic methods, there is a need to develop environmentally benign methodologies for synthesis of benzimidazoles and benzothiazoles.
We report herein an efficient environmentally friendly method for synthesis of benzimidazoles and benzothiazoles in presence of sulfonic-acid-functionalized activated carbon made from matured tea leaf (MTLAC–SA). Sulfonic-acid-functionalized montmorillonite K10 (K10–SA) was also used as catalyst for comparison. Although both catalysts were found to be efficient for synthesis of these heterocycles, MTLAC–SA showed better catalytic activity than K10–SA.
Experimental
Materials and apparatus
The Brunauer–Emmett–Teller (BET) surface area of all the synthesized materials was determined from the N2 adsorption–desorption isotherm at −196 °C using a surface area analyzer (Beckmann Coulter SA-3100). Scanning electron microscopy (SEM) and energy-dispersive X-ray analysis (EDX) of the synthesized materials were carried out using a JEOL JSM-6360 scanning electron microscope. Fourier-transform infrared (FT-IR) spectra were recorded on a Shimadzu IRAffinity-1 spectrophotometer. 1H nuclear magnetic resonance (NMR) spectra were obtained in CDCl3 and dimethylsulfoxide (DMSO) using a Bruker 300 MHz instrument.
Synthesis of catalysts
Synthesis of activated carbon (MTLAC)
Activated carbon was synthesized by following a previously reported procedure [34]. The raw material, i.e., matured tea leaf (MTL), for preparation of activated carbon, was collected from a tea garden of Upper Assam. MTL was washed thoroughly with distilled water to remove adhering dust and impurities, dried, ground, and sieved to obtain 60-mesh-size particles. MTL powder was soaked in 85% H3PO4 at ratio of 1:3 (MTL:H3PO4, w/v) and kept in an oven for 3 h at 60 °C. Acid-impregnated MTL was carbonized in a tube furnace under nitrogen atmosphere. It was heated from room temperature to 500 °C (at heating rate of 5 °C/min) with 1 h holding time at 100 °C intervals. After cooling to room temperature, the activated carbon was washed thoroughly with distilled water until the washing water became neutral, then dried in oven at 100 °C for about 8 h. The resulting activated carbon (MTLAC) was ground, sieved, and kept in a desiccator for further use.
Synthesis of sulfonic-acid-functionalized MTLAC and K10
Sulfonic-acid-functionalized activated carbon was prepared as follows [35]:
Initially, MTLAC powder (200 mg) was dispersed in 5 mL dry CH2Cl2 by ultrasonic bath for 30 min. Chlorosulfonic acid (0.2 mL) was added dropwise to an ice-cooled solution of MTLAC over a period of 30 min at room temperature. After completion of the addition, the mixture was stirred for another 3 h for complete dissipation of HCl from the reaction vessel. The resulting product was filtered and washed with ethanol and water and finally dried in an oven at 80 °C to obtain sulfonic-acid-functionalized activated carbon (MTLAC–SA). Sulfonic-acid-functionalized montmorillonite K10 was prepared by the same procedure as above and is denoted as K10–SA.
General procedure for synthesis of benzimidazole and benzothiazole derivatives
Aldehyde (1 mmol), ortho-phenylenediamine or 2-aminothiophenol (1.1 mmol), synthesized catalyst (10 wt%), and ethanol (3 mL) were mixed and stirred at room temperature for appropriate time. After reaction completion as confirmed by thin-layer chromatography (TLC), the usual work-up procedure was carried out and the crude product was purified using column chromatography to obtain the pure product. Both the catalysts were recovered after reaction, washed with ethanol, dried in an oven for about 1 h, and reused for seven cycles successfully.
Experimental data for synthesized benzimidazole and benzothiazole derivatives
2-Phenyl-1H-benzimidazole (1a)
M.p. 287–290 °C (lit. 290–291 °C) [36]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.18–7.20 (m, 2H), 7.47–7.64 (m, 5H), 8.16 (d, J = 7.8 Hz, 2H).
2-p-Tolyl-1H-benzimidazole (1b)
M.p. 261–264 °C (lit. 262–265 °C) [36]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 2.33 (s, 3H), 7.18–7.23 (m, 4H), 7.59–7.62 (m, 2H), 8.05 (d, J = 8.1 Hz, 2H).
2-(4-Methoxyphenyl)-1H-benzimidazole (1c)
M.p. 224–226 °C (lit. 225–226 °C) [36]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 3.76 (s, 3H), 6.90 (d, J = 8.7 Hz, 2H), 7.13–7.16 (m, 2H), 7.53–7.56 (m, 2H), 8.09 (d, J = 8.7 Hz, 2H).
2-(2-Methoxyphenyl)-1H-benzimidazole (1d)
M.p. 163–165 °C (lit. 157–158 °C) [37]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 3.99 (s, 3H), 6.97–7.06 (m, 2H), 7.16–7.19 (m, 2H), 7.36 (t, J = 8.7 Hz, 1H), 7.60–7.64 (m, 2H), 8.45–8.47 (d, J = 6 Hz, 1H).
2-(3,4-Dimethoxyphenyl)-1H-benzimidazole (1e)
M.p. 186–188 °C (lit. 180–181 °C) [38]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 3.86 (s, 6H), 6.87 (d, J = 8.4 Hz, 1H), 7.16–7.20 (m, 2H), 7.56–7.59 (m, 2H), 7.71 (d, J = 8.1 Hz, 1H), 7.79 (s, 1H).
2-(4-Chlorophenyl)-1H-benzimidazole (1f)
M.p. 286–288 °C (lit. 288–290 °C) [36]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.22 (s, 2H), 7.62 (d, J = 7.8 Hz, 4H), 8.17 (d, J = 7.5 Hz, 2H).
2-(4-Fluorophenyl)-1H-benzimidazole (1g)
M.p. 248–252 °C (lit. 251 °C) [17]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.08 (t, J = 8.7 Hz, 2H), 7.18–7.21 (m, 2H), 7.57–7.60 (m, 2H), 8.14–8.19 (m, 2H).
2-(2-Fluorophenyl)-1H-benzimidazole (1h)
M.p. 223–225 °C (lit. 232–235 °C) [39]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.20–7.25 (m, 1H), 7.30–7.36 (m, 3H), 7.45 (t, J = 7.5 Hz, 1H), 7.52 (s, 1H), 7.86 (s, 1H), 8.52 (t, J = 7.5 Hz, 1H), 10.02 (s, 1H).
2-(4-Bromophenyl)-1H-benzimidazole (1i)
M.p. 295–298 °C (lit. 294–296 °C) [17]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.12–7.15 (m, 2H), 7.51 (d, J = 8.7 Hz, 4H), 7.99 (d, J = 8.7 Hz, 2H).
2-(4-Nitrophenyl)-1H-benzimidazole (1j)
M.p. 317–320 °C (lit. 319–321 °C) [36]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.19–7.22 (m, 2H), 7.50–7.57 (m, 2H), 8.26 (t, J = 7.8 Hz, 2H), 8.37 (d, J = 8.7 Hz, 2H).
2-(3-Nitrophenyl)-1H-benzimidazole (1k)
M.p. 196–199 °C (lit. 203–205 °C) [40]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.38–7.24 (m, 2H), 7.65 (t, J = 7.8 Hz, 3H), 8.24 (d, J = 7.8 Hz, 1H), 8.63 (d, J = 7.5 Hz, 1H), 9.09 (s, 1H).
2-Heptyl-1H-benzimidazole (1l)
M.p. 134–138 °C (lit. 138–140 °C) [38]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 0.80–0.87 (m, 3H), 1.21–1.35 (m, 8H), 1.82–1.89 (m, 2H), 2.99 (t, J = 8.1 Hz, 2H), 6.16 (s, 1H), 7.23–7.27 (m, 2H), 7.60 (s, 2H).
2-(1H-pyrrol-2-yl)-1H-benzimidazole (1m)
M.p. 275–277 °C (lit. 273–275 °C) [37]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 6.11 (t, J = 5.4 Hz, 1H), 6.87 (s, 1H), 6.93 (d, J = 3 Hz, 1H), 7.04–7.08 (m, 2H), 7.36–7.40 (m, 2H), 11.59 (s, 1H).
2-(Anthracen-9-yl)-1H-benzimidazole (1n)
M.p. 255 °C (lit. 261–263 °C) [41]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.33–7.44 (m, 6H), 7.61 (d, J = 7.8 Hz, 2H), 7.72–7.75 (m, 2H), 7.98 (d, J = 8.4 Hz, 2H), 8.52 (s, 1H).
2-(Naphthalen-1-yl)-1H-benzimidazole (1o)
M.p. 205–207 °C (lit. 212–215 °C) [38]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.08–7.10 (m, 2H), 7.35 (s, 2H), 7.52 (s, 2H), 7.68 (d, J = 4.2 Hz, 1H), 7.76 (d, J = 8.1 Hz, 2H), 8.15 (d, J = 8.4 Hz, 1H), 8.58 (s, 1H).
2-(Furan-2-yl)-1H-benzimidazole (1p)
M.p. 284–285 °C (lit. 278–283 °C) [38]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 6.44–6.46 (m, 1H), 7.10–7.13 (m, 2H), 7.26 (t, J = 3.3 Hz, 1H), 7.46–7.50 (m, 3H).
2-Styryl-1H-benzimidazole (1q)
M.p. 200–204 °C (lit. 200–202 °C) [17]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 7.04–6.96 (m, 3H), 7.11–7.18 (m, 3H), 7.33–7.40 (m, 4H), 7.57 (d, J = 16.8 Hz, 1H).
4-(1H-Benzimidazol-2-yl) benzene-1,2-diol (1r)
M.p. 255–257 °C (lit. 258 °C) [42]; 1H NMR (300 MHz, CDCl3 + DMSO-d 6 ): δ (ppm): 6.69 (d, J = 8.4 Hz, 1H), 6.90–6.93 (m, 2H), 7.30–7.33 (m, 3H), 7.46 (s, 1H).
5,6-Dichloro-2-p-tolyl-1H-benzimidazole (1s)
M.p. 220–221 °C (lit. 223–225 °C) [43]; 1H NMR (300 MHz, DMSO-d 6 ): δ (ppm): 2.37 (s, 3H), 7.36 (d, J = 7.8 Hz, 2H), 7.81 (s, 2H), 8.03 (d, J = 6 Hz, 2H).
6-Methyl-2-p-tolyl-1H-benzimidazole (1t)
M.p. 104–105 °C (lit. 101–103 °C) [44]; 1H NMR (300 MHz, CDCl3): δ (ppm): 2.27 (s, 3H), 2.39 (s, 3H), 7.07–7.00 (m, 3H), 7.33 (s, 1H), 7.46 (d, J = 8.1 Hz, 2H), 8.07 (d, J = 7.8 Hz, 2H).
2-Phenylbenzothiazole (2a)
M.p. 112–115 °C (lit. 111–112 °C) [36]; 1H NMR (300 MHz, CDCl3): δ (ppm): 6.69 (d, J = 8.4 Hz, 1H), 6.90–6.93 (m, 2H), 7.30–7.33 (m, 3H), 7.46 (s, 1H).
2-p-Tolylbenzothiazole (2b)
M.p. 82–86 °C (lit. 81–83 °C) [36]; 1H NMR (300 MHz, CDCl3): δ (ppm): 2.43 (s, 3H), 7.30–7.41 (m, 3H), 7.50 (t, J = 7.8 Hz, 1H), 7.90 (d, J = 7.5 Hz, 1H), 7.99 (d, J = 7.8 Hz, 2H), 8.07 (d, J = 8.4 Hz, 1H).
2-(4-Methoxyphenyl)benzothiazole (2c)
M.p. 125 °C (lit. 120–121 °C) [36]; 1H NMR (300 MHz, CDCl3): δ (ppm): 3.90 (s, 3H), 7.02 (d, J = 9 Hz, 2H), 7.38 (t, J = 7.8 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 8.07 (d, J = 8.7 Hz, 3H).
2-(2-Methoxyphenyl)benzothiazole (2d)
M.p. 108 °C (lit. 110–112 °C) [45]; 1H NMR (300 MHz, CDCl3): δ (ppm): 4.08 (s, 3H), 7.07–7.18 (m, 2H), 7.37–7.42 (m, 1H), 7.45–7.53 (m, 2H), 7.94 (d, J = 8.1 Hz, 1H), 8.13 (d, J = 7.8 Hz, 1H), 8.56 (d, J = 9.6 Hz, 1H).
2-(4-Fluorophenyl)benzothiazole (2e)
M.p. 100 °C (lit. 100–102 °C) [46]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.20 (t, J = 8.7 Hz, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.07–8.13 (m, 3H).
2-(2-Fluorophenyl)benzothiazole (2f)
M.p. 91–93 °C (lit. 95–98 °C) [39]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.23–7.37 (m, 2H), 7.42–7.58 (m, 3H), 7.96 (d, J = 8.1 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.50 (t, J = 8.1 Hz, 1H).
2-(2,4-Dichlorophenyl)benzothiazole (2g)
M.p. 148 °C (lit. 151–152 °C) [45]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.40–7.48 (m, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.97 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H).
2-(3-Nitrophenyl)benzothiazole (2h)
M.p. 190 °C (lit. 194–197 °C) [45]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.46 (t, J = 7.8 Hz, 1H), 7.56 (t, J = 8.1 Hz, 1H), 7.70 (t, J = 8.1 Hz, 1H), 7.96 (d, J = 7.5 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 8.35 (d, J = 9.3 Hz, 1H), 8.43 (d, J = 7.5 Hz, 1H), 8.95 (s, 1H).
2-(4-Bromophenyl)benzothiazole (2i)
M.p. 100 °C (lit. 101–102 °C) [36]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.42 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.90–7.98 (m, 3H), 8.08 (d, J = 8.1 Hz, 1H).
2-Heptyl-1H-benzothiazole (2j)
1H NMR (300 MHz, CDCl3): δ (ppm): 0.88 (t, J = 6.6 Hz, 3H), 1.29–1.48 (m, 8H), 1.82–1.92 (m, 2H), 3.11 (t, J = 9 Hz, 2H), 7.34 (t, J = 7.5 Hz, 1H), 7.45 (t, J = 7.2 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.98 (d, J = 8.1 Hz, 1H).
2-(Naphthalen-1-yl)benzothiazole (2k)
M.p. 126–128 °C (lit. 128–130 °C) [46]; 1H NMR (300 MHz, CDCl3): δ (ppm): 7.42 (t, J = 7.5 Hz, 1H), 7.50–7.58 (m, 3H), 7.88–8.00 (m, 4H), 8.13 (d, J = 8.1 Hz, 1H), 8.23 (d, J = 8.1 Hz, 1H), 8.58 (s, 1H).
Results and discussion
Synthesis of catalysts
Initially, activated carbon was synthesized by following a procedure reported by us [34]. After ensuring formation of activated carbon from matured tea leaves, the surface of activated carbon was further modified by incorporating sulfonic acid functionality on the surface by using chlorosulfonic acid in DCM (Fig. 1).

Schematic representation of the process of synthesis of catalysts
For surface modification, carbon sample was taken in DCM and kept under sonication for 30 min to produce homogeneous dispersion. The dispersion was further kept on an ice bath, and appropriate amount of chlorosulfonic acid was added slowly dropwise. To incorporate maximum amount of sulfonic acid functionality on the carbon surface, a series of experiments was conducted using different amounts of chlorosulfonic acid. It was found that a maximum of 1 mL chlorosulfonic acid per gram of sample could be used for surface modification. Beyond this amount, the carbon sample was converted to a sticky mass after chlorosulfonic acid treatment. For comparison of the catalytic activity, commercially available montmorillonite K10 sample was also modified by incorporating sulfonic acid functionality. It is noteworthy that montmorillonite K10 has already been exploited as catalyst for this particular transformation.
The BET surface area of all the synthesized materials was determined from N2 adsorption–desorption isotherms at − 196 °C. The surface texture and elemental content of the synthesized materials were characterized by scanning electron microscopy (SEM) and energy-dispersive X-ray (EDX) spectroscopic analysis. To confirm the functional groups present in the samples, Fourier-transform infrared (FT-IR) spectra were recorded in the scanning range from 4000 to 400 cm−1. Boehm titration was carried out to determine the total surface acidity of the synthesized catalysts.
BET analysis
The surface area of the synthesized materials was determined by nitrogen adsorption–desorption method using the BET equation. Prior to analysis, the samples were dried at 100 °C then degassed under vacuum at 120 °C for 2 h. The surface area of the materials is presented in Table 1, and the nitrogen adsorption–desorption isotherms are shown in Fig. 2. All the isotherms were found to be of type II as per International Union of Pure and Applied Chemistry (IUPAC) classification. From Table 1, it is seen that MTLAC had higher surface area than K10. After surface functionalization, there was a decrease in surface area for all the materials. Such decrease in surface area may be due to introduction of functional groups onto surface-active sites of the parent material.

N2 adsorption–desorption isotherms of synthesized materials
SEM and EDX analysis
The surface morphology of MTLAC–SA and K10–SA was studied by SEM (Fig. 3). The samples were coated with a thin layer of gold using a sputter coater prior to analysis. As shown in the figure, MTLAC–SA had porous surface, in good agreement with its higher BET surface area among the two catalysts.

Scanning electron micrographs of a MTLAC, b MTLAC–SA, c K10, and d K10–SA
Energy-dispersive X-ray (EDX) spectroscopic analysis was carried out for in situ chemical analysis of the synthesized materials; the results are presented in Tables 2 and 3. Presence of one new element (S) in MTLAC–SA and K10–SA confirmed successful functionalization of sulfonic acid group on the surface of the synthesized materials.
FT-IR analysis
FT-IR spectra of all the synthesized materials are presented in Fig. 3. The signal observed at around 620 cm−1 can be attributed to presence of S–O stretching vibration. The signal observed at around 1120 cm−1 is due to presence of S=O symmetric stretching vibration. The broad band at around 3400 cm−1 can be assigned to S–OH stretching vibration (Fig. 4).

FT-IR spectra of synthesized materials
Boehm titration
Boehm titration was carried out to determine the amount of acidic surface groups on the synthesized materials [47]. Synthesized materials (0.15 g) were mixed with 30 mL 0.05 N NaOH solution. The samples were sealed and shaken for 24 h, then 10 mL filtrate was titrated with 0.05 N HCl. The number of acidic surface groups was calculated based on the assumption that NaOH neutralizes acidic groups present on the material surface. The results of the experiment are presented in Table 4, revealing that MTLAC–SA had slightly more acidic surface groups than K10–SA.
Optimization of reaction conditions
The reaction of 4-methylbenzaldehyde (1 mmol) and ortho-phenylenediamine (1.1 mmol) was selected as model reaction to establish the best reaction conditions. The reaction was first performed without any catalyst for about 5 h at room temperature, obtaining trace (8%) yield. In presence of the synthesized catalyst (MTLAC–SA), 92% yield was obtained in 45 min.
The effect of different solvent systems for synthesis of imidazole derivatives was studied; the results are summarized in Table 5. The reaction was carried out using solvents EtOH, MeOH, water, and acetonitrile. EtOH and acetonitrile showed almost equal results regarding yield of product. Considering the environmental effect, EtOH was chosen as a suitable solvent for the reaction. The model reaction was also optimized in terms of catalyst concentration. The results are summarized in Table 5. In absence of catalyst, the reaction did not produce good yield. However, addition of 5 wt% (with respect to the weight of aldehyde) catalyst resulted in a sharp increase of yield within a short period of time. Addition of 10 wt% catalyst was found to be sufficient for the reaction. Further increase in the amount of catalyst up to 15 wt% did not show any effective increase in product yield.

These optimum conditions were also used for synthesis of benzothiazole derivatives.
To compare the catalytic activity of MTLAC–SA with that of K10–SA, the reaction between 4-methylbenzaldehyde and ortho-phenylenediamine or ortho-aminothiophenol was carried out using both catalysts. The results obtained are depicted in Fig. 5.

Comparison of catalytic activity of MTLAC–SA and K10–SA
From this figure, it is observed that MTLAC–SA showed better catalytic activity in comparison with K10–SA. This can be attributed to the acid capacity of the studied catalyst. According to the Boehm titration results (Table 4), the acid capacity of MTLAC–SA was higher than that of K10–SA. Also, the higher surface area of the catalyst (Table 1) provides a large number of active sites. These are the two major reasons for the superior catalytic performance of MTLAC–SA to that of K10–SA.
After optimizing the reaction conditions, the versatility of this method was examined by performing the reaction using different substituted aldehydes. Aldehydes with both electron-withdrawing and electron-donating substituent were used successfully for synthesis of corresponding imidazole and thiazole derivatives in excellent yield. The results are presented in Table 6. Using the same optimum condition, benzothiazole derivatives were also synthesized; the results obtained are summarized in Table 6. From this table one can say that the catalyst is very effective for benzothiazole synthesis as well.
The effectiveness and applicability of the synthesized catalysts were compared with some reported catalysts in terms of reaction time, percentage yield, and reaction temperature. For this study, synthesis of 2-(4-methoxyphenyl)-1H-benzimidazole and 2-(4-methoxyphenyl)benzothiazole were taken as model reactions (Table 7).
Recyclability of catalysts
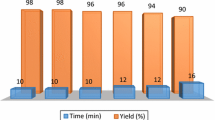
To check the recyclability of the catalysts, the reaction between 4-methylbenzaldehyde and ortho-phenylenediamine was carried out using both catalysts.
After the reaction, the catalysts were separated from the reaction mixture, washed with ethanol, then dried. The recovered catalysts were further used for the same reaction to check the catalytic activity. All the catalysts were successfully used for seven catalytic cycles without much decrease in product yield, as shown in Fig. 6.

Recyclability of both catalysts
Conclusions
An efficient, simple, and green method was developed for synthesis of benzimidazole and benzothiazole derivatives using sulfonic-acid-modified activated carbon (MTLAC–SA) as very effective and recyclable catalyst. The catalytic activity of MTLAC–SA was found to be superior to that of a catalyst prepared by modification of montmorillonite K10 (K10–SA). The synthesis procedure for both catalysts was very simple and cost effective. Short reaction time, good to excellent yield of product, room-temperature condition, and use of inexpensive and reusable catalysts make this method both economically and environmentally suitable.
References
M. Gaba, S. Singh, C. Mohan, Eur. J. Med. Chem. 76, 494 (2014)
M.J. Tebbe, W.A. Spitzer, F. Victor, S.C. Miller, C.C. Lee, T.R. Sattelberg Sr., E. Mckinney, J.C. Tang, J. Med. Chem. 40, 3937 (1997)
V.K. Vyas, M. Ghate, Mini-Rev. Med. Chem. 10, 1366 (2010)
A. Mavrova, K.K. Anichina, D.I. Vuchev, J.A. Tsenov, P.S. Denkova, M.S. Kondeva, M.K. Micheva, Eur. J. Med. Chem. 41, 1412 (2006)
N.C. Desai, N.R. Shihory, G.M. Kotadiya, P. Desai, Eur. J. Med. Chem. 82, 480 (2014)
V. Zaharia, A. Ignat, N. Palibroda, B. Ngameni, V. Kuete, C.N. Fokunang, M.L. Moungang, B.T. Ngadjui, Eur. J. Med. Chem. 45, 5080 (2010)
A. Kamal, M.N.A. Khan, K.S. Reddy, K. Rohini, Bioorg. Med. Chem. 15, 1004 (2007)
F. Azam, B.A. El-Gnidi, I.A. Alkskas, M.A. Ahmed, J. Enzyme Inhib. Med. Chem. 25, 818 (2010)
A.R. Porcari, R.V. Devivar, L.S. Kucera, J.C. Drach, L.B. Townsend, J. Med. Chem. 41, 1252 (1998)
M.T. Migawa, J.L. Giradet, J.A. Walker, G.W. Koszalka, S.D. Chamber-Lain, J.C. Drach, L.B. Townsend, J. Med. Chem. 41, 1242 (1998)
W.A. Denny, G.W. Rewcastle, B.C. Bagley, J. Med. Chem. 33, 814 (1990)
T. Forseca, B. Gigante, T.L. Gilchrist, Tetrahedron 57, 1793 (2001)
S. Aiello, G. Wells, E.L. Stone, H. Kadri, R. Bazzi, D.R. Bell, M.F.G. Stevens, C.S. Matthews, T.D. Bradshaw, A.D. Westwell, J. Med. Chem. 51, 5135 (2008)
S.J. Choi, H.J. Park, S.K. Lee, S.W. Kim, G. Han, H.Y.P. Choo, Bioorg. Med. Chem. 14, 1229 (2006)
D. Alagille, R.M. Baldwin, G.D. Tamagnan, Tetrahedron Lett. 46, 1349 (2005)
S.J. Ji, H.B. Shi, Dyes Pigm. 70, 246 (2006)
J. Azizian, P. Torabi, J. Noei, Tetrahedron Lett. 57, 185 (2016)
A. Dandia, V. Parewa, K.S. Rathore, Catal. Comm. 28, 90 (2012)
R. Shelkar, S. Sarode, J. Nagarkar, Tetrahedron Lett. 54, 6986 (2013)
Z.H. Zhang, J.J. Li, Y.Z. Gao, Y.H. Liu, J. Heterocycl. Chem. 44, 1509 (2007)
D. Villemin, M. Hammadi, B. Martin, Synth. Commun. 26, 2895 (1996)
Y. Wang, K. Sarris, D.R. Sauer, S.W. Djuric, Tetrahedron Lett. 47, 4823 (2006)
A.K. Chakraborti, C. Selvam, G. Kaur, S. Bhagat, Synlett 5, 0851 (2004)
X. Wen, J.E. Bakali, R. Deprez-Poulain, B. Deprez, Tetrahedron Lett. 53, 2440 (2012)
G.M. Raghavendra, A.B. Ramesha, C.N. Revanna, K.N. Nandeesh, K. Mantelingu, K.S. Rangappa, Tetrahedron Lett. 52, 5571 (2011)
R.N. Nadaf, S.A. Siddiqui, T. Daniel, R.J. Lahoti, K.V. Srinivasan, J. Mol. Catal. A: Chem. 214, 155 (2004)
M.S. Mayo, X. Yu, X. Zhou, X. Feng, Y. Yamamoto, M. Bao, Org. Lett. 16, 764 (2014)
M.S. Mayo, X. Yu, X. Zhou, X. Feng, Y. Yamamoto, M. Bao, J. Org. Chem. 79, 6310 (2014)
G.M. Martins, T. Puccinelli, R.A. Gariani, F.R. Xavier, C.C. Silveira, S.R. Mendes, Tetrahedron Lett. 58, 1969 (2017)
F. Shirini, M. Abedini, M. Seddighi, F.S. Arbosara, Res. Chem. Intermed. 41, 7683 (2015)
H.D. Hanoon, E. Kowsari, M. Abdouss, H. Zandi, M.H. Ghasemi, Res. Chem. Intermed. 43, 1751 (2017)
K.B. Dhopte, R.S. Zambare, A.V. Patwardhan, P.R. Parag, RSC Adv. 6, 8164 (2016)
P.L. Reddy, R. Arundhathi, M. Tripathi, D.S. Rawat, RSC Adv. 6, 53596 (2016)
M. Goswami, L. Borah, D. Mahanta, P. Phukan, J. Porous Mater. 21, 1025 (2014)
H. Naeimi, S. Mohamadabadi, Dalton Trans. 43, 12967 (2014)
E. Dezfoolinezhad, K. Ghodrati, R. Badri, New J. Chem. 40, 4575 (2016)
L.-H. Du, X.-P. Luo, Synth. Commun. 40, 2880 (2010)
M.A. Chari, D. Shobha, T. Sasaki, Tetrahedron Lett. 52, 5575 (2011)
Y.-C. Lin, N.-C. Li, Y.-J. Cherng, J. Heterocycl. Chem. 51, 808 (2014)
R. Srinivasulu, K.R. Kumar, P.V.V. Satyanarayana, Green Sustain. Chem. 4, 33 (2014)
D. Saha, A. Saha, B.C. Ranu, Green Chem. 11, 733 (2009)
S.S. Panda, S.C. Jain, Synth. Commun. 41, 729 (2011)
O. Ravi, A. Shaikh, A. Upare, K.K. Singarapu, S.R. Bathula, J. Org. Chem. 82, 4422 (2017)
K. Bahrami, M.M. Khodaei, F. Naali, J. Org. Chem. 73, 6835 (2008)
G.F. Chen, L.Y. Zhang, H.M. Jia, B.H. Chen, J.T. Li, S.X. Wang, G.Y. Bai, Res. Chem. Intermed. 39, 2077 (2013)
M. Chhabra, S. Sinha, S. Banerjee, P. Paira, Bioorg. Med. Chem. Lett. 26, 213 (2016)
H.P. Boehm, Adv. Catal. 16, 179 (1966)
G.F. Chen, N. Xiao, J.S. Yang, H.Y. Li, B.H. Chen, L.F. Han, Res. Chem. Intermed. 41, 5159 (2015)
A. Teimouria, A.N. Chermahinib, H. Salavatia, L. Ghorbanianc, J. Mol. Catal. A: Chem. 373, 38 (2013)
M.A. Chari, D. Shobha, E.L.-R. Kenawy, S.S. Al-Deyab, B.V. Subba Reddy, A. Vinu, Tetrahedron Lett. 51, 5195 (2010)
G. Brahmachari, S. Laskar, P. Barik, RSC Adv. 3, 14245 (2013)
M.A. Chari, P. Sadanandam, D. Shobha, K. Mukkanti, J. Heterocycl. Chem. 47, 153 (2010)
Acknowledgements
Financial support from DST [grant no. SR/NM/NS-18/2011(G)] is gratefully acknowledged. M.M.D. thanks UGC for a research fellowship. We also acknowledge support from the Department of Chemistry, Gauhati University, SAIF-GU, SAIF-NEHU, and CIF-IITG for all the analytical facilities used during the course of this investigation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Goswami, M., Dutta, M.M. & Phukan, P. Sulfonic-acid-functionalized activated carbon made from tea leaves as green catalyst for synthesis of 2-substituted benzimidazole and benzothiazole. Res Chem Intermed 44, 1597–1615 (2018). https://doi.org/10.1007/s11164-017-3187-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3187-x