
Abstract
We report synthesis of silica nanospheres containing ferrocene-tagged imidazolium acetate (SiO2@Im-Fc[OAc]) as efficient heterogeneous nanocatalyst for synthesis of naphthopyran derivatives under solvent-free conditions, based on modification of nano SiO2 by ionic liquid with ferrocene tags and subsequent anion metathesis reaction. The synthesized novel nanocatalyst (SiO2@Im-Fc[OAc]) was systematically characterized using Fourier-transform infrared spectroscopy, energy-dispersive X-ray spectroscopy, X-ray diffraction analysis, and field-emission scanning electron microscopy. The catalytic activity of (SiO2@Im-Fc[OAc]) was tested in one-pot three-component reaction of aromatic aldehydes, malononitrile, and 2-naphthol for facile synthesis of naphthopyran derivatives. To achieve high catalytic efficacy, the effects of various reaction parameters such as temperature, amount of catalyst, type of solvent, etc. were investigated. Furthermore, recovery and reuse of the nanocatalyst several times was demonstrated without appreciable loss in catalytic activity. The presented protocol offers several advantages, including green and ecofriendly nature, operational simplicity, higher yield, and easy recovery and reuse of the nanostructured catalyst. The workup of these very clean reactions involves only recrystallization of the product from ethanol and recovery of the catalyst by filtration.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, achieving “green” chemistry principles using cleaner, ecofriendly, and benign synthetic processes has become one of the main aims in organic synthesis [1]. In this context, several techniques for efficient use of solvent-free reactions [2], multicomponent reactions [3], and reusable heterogeneously catalyzed reactions [4] have been developed individually, but when these three aspects of green chemistry can be combined, excellent green chemistry protocols can be expected [5].
Ionic liquids represent another new environmentally friendly approach to green chemistry, finding application in a wide range of areas, especially catalysis of organic reactions [6]. Furthermore, task-specific ionic liquids (TSILs) have received more attention because of their potential application in replacing conventional homogeneous/heterogeneous catalysts [7,8,9]. Because the structural and chemical versatility of ILs make them appropriate candidates for functionalization of solid materials [10], supported ionic liquids (SILs) have been prepared by immobilizing ILs on solid materials [11]. In this regard, use of silica-supported ionic liquids as new catalysts is of considerable importance due to their enhanced reaction rate, facile catalyst handling, low cost, simple workup and separation from the reaction mixture, ease of preparation, and recoverability [12]. SILs therefore combine the superior properties of homogeneous ionic liquids, nanomaterials, and heterogeneous solids [13, 14]. Against this background, development and expansion of new nanocatalysts by immobilization of TSILs on nanoparticles (NPs) needs more attention and research.
Pyrans are an important class of heterocycles that display remarkable biological and pharmacological activities. These heterocycles are also found in bioactive natural heterocyclic compounds with pharmaceutical potential [15,16,17]. Compounds having a pyran skeleton possess varied pharmacological activities such as antibacterial [18], antitumor [19], and anti-human immunodeficiency virus (HIV) [20] actions. These compounds have been found useful for treatment of Alzheimer’s, schizophrenia, and myoclonus diseases [21]. In this regard, naturally occurring naphthopyrans have a wide range of interesting physiological properties and biological activities [22]. Naphthopyrans also show antiproliferative [23], cytotoxic [24], mutagenic, and anticancer [25] activities. They also include an important class of photochromic compounds with a wide variety of applications [26]. Therefore, synthesis of such compounds has attracted strong interest [27]. Generally, synthesis of naphthopyran derivatives is performed by cyclization of malononitrile, aldehyde, and naphthol [27]. Various catalysts have been reported for synthesis of these compounds, including 1,4-diazabicyclo[2.2.2]octane (DABCO) [28], ZnAl2O4–Bi2O3 [29], acetic acid ionic liquid [30], silica-bonded aminoethylpiperazine [31], Ni–Al2O3 [32], and MgO [33], but most of them are associated with one or more drawbacks, such as long reaction time, harsh conditions, toxic organic solvents, difficult workup procedures, and multistep reaction conditions with reduced yield. So, development of efficient methods that can be conducted under solvent-free conditions employing environmentally benign catalysts is still a valid goal.
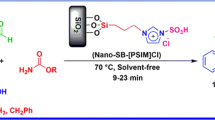
In continuation of our research on synthesis and applications of ferrocene derivatives [34, 35] and modification and development of multicomponent reactions [36,37,38], we report herein a ferrocene-containing imidazole-based ionic liquid stabilized on silica nanoparticles (SiO2@Im-Fc[OAc]) as efficient catalyst for synthesis of a series of naphthopyran derivatives under solvent-free conditions (Scheme 1).

Synthesis of naphthopyran derivatives using (SiO2@Im-Fc[OAc]) as novel catalyst
Results and discussion
This research was performed in two stages. Initially, nanosilica was prepared and subsequently modified by ferrocene-tagged ionic liquid (Scheme 2). In the next step, (SiO2@Im-Fc[OAc]) nanocatalyst was used for synthesis of naphthopyran derivatives under solvent-free condition.

Preparation steps for synthesis of (SiO2@Im-Fc[OAc]) nanocatalyst
Synthesis and characterization of (SiO2@Im-Fc[OAc]) nanocatalyst
Nanosilica-supported ferrocene-containing ionic liquid (SiO2@Im-Fc[OAc]) as nanocatalyst was synthesized via a multistep reaction as shown in Scheme 2. First, silica nanoparticle (SNP) shells were made by a liquid chemical approach based on the well-known Stöber process, which involves hydrolysis and condensation of tetraethylorthosilicate (TEOS) in methanol in presence of water with ammonia as catalyst [39]. Subsequently, the synthesized SNPs were reacted with triethoxy-3-(chloropropyl)-silane in ethanol to obtain propylchloride-functionalized nanosilica. On the other hand, 1-N-ferrocene butylimidazole was prepared by reaction of imidazole with chlorobutyl ferrocene. Then, ferrocene-tagged ionic liquid supported on silica nanoparticles was synthesized by reaction of chloropropyl-modified silica-coated magnetic nanoparticles with 1-(4-ferrocenylbutyl)-1H-imidazole. Finally, reaction of the obtained (SiO2@Im-Fc[Cl]) with NaOAc produced (SiO2@Im-Fc[OAc]). The structure of the synthesized nanocatalyst was studied and fully characterized using Fourier-transform infrared (FT-IR) spectroscopy, energy-dispersive X-ray (EDX) spectroscopy, X-ray diffraction (XRD) analysis, and field-emission scanning electron microscopy (FE-SEM).
The FT-IR results for SiO2@propylchloride, (SiO2@Im-Fc[Cl]), and (SiO2@Im-Fc[OAc]) catalyst are shown in Fig. 1. The broad absorption band at around 1100 cm−1 is related to asymmetric stretching of Si–O–Si. The absorption bands at 802 and 469 cm−1 can be attributed to bending vibration of Si–O–Si bonds. The absorption peaks at 2880 and 2945 cm−1 are attributed to asymmetric and symmetric C–H stretching in propyl and butyl chain. The absorption peak at 3180 cm−1 is also linked with stretching vibration of aromatic C–H on imidazole and ferrocene. The absorption peaks at 1644 and 1385 cm−1 are also correlated to stretching vibration of C=C and C=N bond on aromatic rings. The broad absorption band at around 3435 cm−1 can be related to absorption of –O–H groups on the surface of silica. The absorption peak at 1710 cm−1 is related to stretching vibration of C=O bond of carbonyl group in acetate anion. These results confirm the surface modification of the nano SiO2 and successful synthesis of (SiO2@Im-Fc[OAc]).

FT-IR spectra of a SiO2@propylchloride, b (SiO2@Im-Fc[Cl]), and c (SiO2@Im-Fc[OAc]) nanocatalyst
The energy-dispersive X-ray (EDX) spectra of the (SiO2@Im-Fc[OAc]) catalyst confirmed the presence of the expected elements in the structure of the catalyst with presence of Fe, Cl, Si, O, N, and C in their respective regions (Fig. 2). It is clearly seen that the synthesized nanocatalyst contained only C, N, O, Fe, and Si elements. The absence of chlorine (Cl) from the final nanocatalyst is noteworthy, confirming the completeness of the loading of the acetate anion and metathesis reaction.

EDX results for a (SiO2@Im-Fc[OAc]) and b (SiO2@Im-Fc[Cl]) nanocatalyst
The XRD patterns of the synthesized nano SiO2, (SiO2@Im-Fc[Cl]), and (SiO2@Im-Fc[OAc]) catalyst are shown in Fig. 3. A single broad diffraction peak is observed at 2θ = 23°, being characteristic of the amorphous nature of Stöber’s silica nanospheres. The results show that no remarkable change was observed in the XRD pattern of the silica nanospheres even after surface functionalization reactions.

XRD patterns of a SiO2, b (SiO2@Im-Fc[Cl]), and c (SiO2@Im-Fc[OAc]) nanocatalyst
The morphology of the synthesized (SiO2@Im-Fc[OAc]) catalyst was examined by field-emission scanning electron microscopy (FE-SEM). As revealed in Fig. 4, the increased surface roughness of these (SiO2@Im-Fc[OAc]) spheres indicates successful formation of an additional layer around the core SiO2 particles, because Stöber’s silica nanospheres possess a rather smooth surface. Changes in surface roughness will also affect the active surface area and cause higher catalytic activity.

FE-SEM images of (SiO2@Im-Fc[OAc]) nanocatalyst at different magnifications: a 2 µm, b 1 µm, and c 500 nm
Catalytic activity of (SiO2@Im-Fc[OAc])
To optimize the reaction conditions to obtain the best catalytic activity and investigate the feasibility of the strategy, the reaction of 2-naphthol (1a) with benzaldehyde (2a) and malononitrile (3a) was chosen as model reaction. For this purpose, we investigated the efficiency of (SiO2@Im-Fc[OAc]) using a variety of solvents, different temperatures, and different amounts of synthesized nanocatalyst to identify a more effective, simple, and rapid method for synthesis of naphthopyran derivatives. The results are summarized in Table 1.

First, a variety of conventional organic solvents such as EtOH, CH2Cl2, CHCl3, CH3CN, and H2O and solvent-free condition were screened for the model reaction at room temperature in presence of 30 mg nanocatalyst (Table 1, entries 1–6). As shown in Table 1, different conventional solvents afforded low to moderate yields at room temperature (18–45 %, entries 1–5). Interestingly, the best result was achieved under solvent-free condition. The model reaction was then carried out in presence of different quantities of nanocatalyst (50, 40, 30, 20, 10, and 5 mg) and at various temperatures. It was observed that excellent yield was achieved when using 10 mg (SiO2@Im-Fc[OAc]) (Table 1, entry 10) and further decrease or increase in the catalyst quantity beyond 10 mg did not increase the yield of the product significantly. As shown in Table 1, the best result (90% yield) was obtained when the reaction was performed using 10 mg catalyst at 90 °C (entry 16). Notably, there was no need for inert atmosphere and the reactions were carried out at ambient conditions.
After optimization of the reaction conditions, condensation reaction of 2-naphthols with a variety of aromatic aldehydes and malononitrile was examined in presence of nanocatalyst (10 mg) at 90 °C under solvent-free condition to explore the ability of the catalyst and generality of the procedure. The results are summarized in Table 2.

Finally, we investigated the reusability of the catalyst under solvent-free conditions using a model reaction in presence of (SiO2@Im-Fc[OAc]) nanocatalyst under the optimized conditions. After completion of the reaction, the reaction mixture was dissolved in hot ethanol and the catalyst was easily separated from the reaction mixture by filtration with no obvious catalyst mass loss observed. The catalyst was washed twice with hot ethanol and dried in vacuum. The recovered catalyst was reused for six runs with only slight decrease in its weight and activity (Fig. 5).

Reusability of (SiO2@Im-Fc[OAc]) nanocatalyst for synthesis of 4a
Conclusions
We designed and synthesized a novel nanosilica ionic liquid based on imidazolium with ferrocene tags and used it as a highly efficient heterogeneous nanocatalyst for direct synthesis of naphthopyran derivatives. A series of naphthopyrans were synthesized via one-pot three-component reaction of various aldehydes, malononitrile, and 2-naphthol in presence of the catalyst under solvent-free condition. The operational simplicity of the procedure, easy workup, and environmental friendliness make this method more attractive. In addition, the catalyst can be easily recovered and recycled for at least six times without loss of efficacy.
Experimental
Materials and apparatus
The chemical reagents used in synthesis were purchased from Merck and Sigma–Aldrich companies. Melting points were determined using a MEL-TEMP model 1202D and are uncorrected. FT-IR spectra were recorded on a Bruker Tensor 27 spectrometer from KBr disks. 1H nuclear magnetic resonance (NMR) spectra were recorded with a Bruker Spectrospin Avance 400 spectrometer with dimethylsulfoxide (DMSO)-d6 as solvent and tetramethylsilane (TMS) as internal standard. 13C NMR spectra were determined on the same instrument at 100 MHz. All chemical shifts are reported as δ (ppm), and coupling constants (J) are given in Hz. Elementary analyses (C, H, N) were performed on a Vario EL III analyzer. Sonication was performed using a Hielscher (UP400s) ultrasonic probe system at frequency of 24 kHz. X-ray diffraction patterns of samples were taken on a Siemens D500 X-ray powder diffractometer (Cu K radiation, λ = 1.5406 Å). FE-SEM images of the products were obtained using a TESCAN MIRA3.
Synthesis of (SiO2@Im-Fc[OAc]) catalyst
Synthesis of SiO2@propylchloride
Initially, monodisperse silica nanospheres (SiO2) were synthesized via Stöber’s method by hydrolysis of tetraethylorthosilicate (TEOS) using ammonium hydroxide in ethanol [39]. In a typical synthesis, TEOS (1 mL) was added into 10 mL ethanol, then ammonium hydroxide 25 % (10 mL) and 10 mL ethanol were also introduced to the reaction mixture under sonication. After 60 min, white suspension of silica was produced, then the reaction mixture was centrifuged to separate silica nanospheres. The precipitate was washed with water and ethanol, respectively. Then, silica nanospheres were functionalized with propylchloride by addition of 3-chloropropyltriethoxysilane (1 mL) to a vigorously stirred dispersion of SiO2 in ethanol (50 mL), and the mixture was refluxed for 24 h. The resulting propylchloride-functionalized silica nanospheres (SiO2@propylchloride) were separated by centrifugation and redispersed in ethanol, which was repeated three times.
Synthesis of 1-(4-ferrocenylbutyl)-1H-imidazole
To suspension of NaH 60 % (0.8 g, 20 mmol) in dry tetrahydrofuran (THF) (100 mL), imidazole (1.36 g, 20 mmol) was added and stirred for 1 h at 0 °C. Then, 4-chlorobutylferrocene (1.38 g, 5 mmol) was added and refluxed for 12 h. The reaction mixture was cooled to 0 °C, and excess NaH was quenched by addition of small amount of water. The reaction mixture was poured into water and extracted with CH2Cl2. The combined extracts were dried with MgSO4, and the solvent was removed under reduced pressure. The residue was purified by silica column chromatography (n-hexane/ethyl acetate, 9:1 v/v) to give 1-(4-ferrocenylbutyl)-1H-imidazole as brown viscous oil. FT-IR (KBr): 3394, 2859, 1506, 1453, 1105, 1228, 1000, 650, 486 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.42–1.49 (m, 2H, –CH2–), 1.75–1.78 (m, 2H, –CH2–), 2.34 (t, J = 7.5 Hz, 2H, –CH2–), 3.90 (t, J = 6.6 Hz, 2H,–CH2), 4.00–4.03 (m, 4H, Cp-H), 4.06 (s, 5H, Cp-H), 6.98–7.26 (m, 2H, imid-H), 7. 49 (s, 1H, imid-H) ppm; 13C NMR (100 MHz, CDCl3): δ 27.0, 28.0, 29.7, 45.9, 66.1, 66.9, 67.3, 87.0, 118.2, 128.7, 136.6 ppm.
Synthesis of (SiO2@Im-Fc[Cl])
The mixture of SiO2@propylchloride and 1-N-ferrocenyl butylimidazole was stirred for 72 h in toluene solution at 80 °C, and the obtained (SiO2@Im-Fc[Cl]) was filtered, washed with toluene (3 × 20 mL) and CH2Cl2 (3 × 20 mL), then dried under vacuum at 50 °C for 48 h.
Synthesis of (SiO2@Im-Fc[OAc])
A mixture of (SiO2@Im-Fc[Cl]) (1.0 g) and sodium acetate (2 g) in 30 mL water was stirred at room temperature for ion exchange. After 24 h, the residue was filtered, washed with with water (3 × 40 mL) and acetone (2 × 30 mL), and dried under vacuum at 50 °C for 48 h to afford (SiO2@Im-Fc[OAc]).
General procedure for synthesis of naphthopyran derivatives
To mixture of 2-naphthol (1 mmol), aldehyde (1 mmol), and malononitrile (1.2 mmol) was added (SiO2@Im-Fc[OAc]) (10 mg). The mixture was stirred at 90 °C under solvent-free condition in an oil bath, and reaction completion was monitored by thin-layer chromatography (TLC) (EtOAc/n-hexane: 1/4). After completion of the reaction, the reaction mixture was dissolved in hot ethanol; the catalyst was recovered by filtration, and the product was recrystallized from ethanol to give corresponding naphthopyran as pure solid. The structures of all the new products were confirmed by FT-IR, 1H NMR, 13C NMR, and CHNS analyses.
Spectral data of new compounds
3-Amino-1-(thiophen-2-yl)-1H-benzo[f]chromene-2-carbonitrile ( 4j )
White solid; m.p. 258–260 °C; FT-IR (KBr) ν 3441, 3343, 3082, 2874, 2178, 1639, 1587, 1409 cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 5.71 (1H, s, CH), 6.86–6.88 (1H, m, Ar–H), 7.01 (1H, d, J = 2.90 Hz, Ar–H), 7.14 (2H, s, NH2), 7.24–7.26 (1H, m, Ar–H), 7.30 (1H, d, J = 8.93 Hz, Ar–H), 7.42–7.45 (1H, m, Ar–H), 7.48–7.52 (1H, m, Ar–H), 7.90–7.93 (2H, m, Ar–H), 8.03 (1H, d, J = 8.38 Hz, Ar–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 33.17, 57.81, 116.01, 116.78, 120.38, 123.46, 124.05, 124.81, 125.06, 126.66, 127.19, 128.46, 129.64, 130.08, 130.75, 146.33, 150.25, 160.30 ppm; Anal. Calc. for C18H12N2OS (%): C, 71.03; H, 3.97; N, 9.20; S, 10.53; found: C, 71.08; H, 4.12; N, 9.43; S, 10.34.
3-Amino-1-(5-methylfuran-2-yl)-1H-benzo[f]chromene-2-carbonitrile ( 4l )
Brown solid; m.p. 152–156 °C; FT-IR (KBr) ν 3429, 3326, 3068, 2921, 2182, 1644, 1587, 1411 cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 2.07 (3H, s, CH3), 5.41 (1H, s, CH), 5.89 (1H, d, J = 1.95 Hz, Ar–H), 6.10 (1H, d, J = 2.97 Hz, Ar–H), 7.08 (2H, s, NH2), 7.28 (1H, d, J = 8.94 Hz, Ar–H), 7.44–7.47 (1H, m, Ar–H), 7.50–7.54 (1H, m, Ar–H), 7.90–7.93 (2H, m, Ar–H), 8.06 (1H, d, J = 8.41 Hz, Ar–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 13.42, 31.84, 54.43, 106.44, 106.60, 113.77, 116.90, 120.50, 123.39, 125.06, 127.14, 128.45, 129.49, 130.26, 130.66, 146.88, 150.78, 154.33, 160.94 ppm; Anal. Calc. for C19H14N2O2 (%): C, 75.48; H, 4.67; N, 9.27; found: C, 75.72; H, 4.76; N, 9.21.
3-Amino-8-bromo-1-(4-isopropylphenyl)-1H-benzo[f]chromene-2-carbonitrile ( 4n )
Brown solid; m.p. 211–213 °C; FT-IR (KBr) ν 3419, 3354, 3173, 2878, 2187, 1651, 1532, 1417 cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 1.11 (6H, d, J = 6.78 Hz, CH3), 2.73–2.78 (1H, m, CH), 5.25 (1H, s, CH), 6.98 (2H, s, NH2), 7.05–7.13 (4H, m, Ar–H), 7.38 (1H, d, J = 8.98 Hz, Ar–H), 7.53–7.55 (1H, m, Ar–H), 7.79 (1H, d, J = 9.04 Hz, Ar–H), 7.91 (1H, d, J = 8.99 Hz, Ar–H), 8.19 (1H, s, Ar–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 23.73, 23.77, 32.96, 37.49, 57.91, 116.33, 118.15, 120.48, 125.95, 126.67, 126.81, 127.26, 128.66, 128.85, 129.91, 130.25, 132.14, 142.89, 146.68, 147.09, 159.58, 159.62 ppm; Anal. Calc. for C23H19BrN2O (%): C, 65.88; H, 4.57; N, 6.68; found: C, 65.97; H, 4.64; N, 6.59.
3-Amino-8-bromo-1-(4-fluorophenyl)-1H-benzo[f]chromene-2-carbonitrile ( 4o )
White solid; m.p. 258–262 °C; FT-IR (KBr) ν 3455, 3301, 3068, 2867, 2199, 1663, 1590, 1403 cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 5.35 (1H, s, CH), 7.01–7.10 (4H, m, NH2, Ar–H), 7.18–7.21 (2H, m, Ar–H), 7.39 (1H, d, J = 8.94 Hz, Ar–H), 7.53–7.55 (1H, m, Ar–H), 7.75 (1H, d, J = 9.06 Hz, Ar–H), 7.92 (1H, d, J = 9.02 Hz, Ar–H), 8.19 (1H, s, Ar–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 37.06, 57.63, 115.44, 115.65, 115.85, 118.21, 118.23, 120.30, 125.93, 128.79, 128.82, 128.90, 128.93, 129.96, 130.33, 132.19, 141.71, 141.73, 147.10, 159.49, 159.53, 159.57, 159.67, 162.09 ppm; Anal. Calc. for C20H12BrFN2O (%): C, 60.78; H, 3.06; N, 7.09; found: C, 60.71; H, 2.97; N, 6.91.
3-Amino-8-bromo-1-(3-bromophenyl)-1H-benzo[f]chromene-2-carbonitrile ( 4p )
Light-brown solid; m.p. 198–201 °C; FT-IR (KBr) ν 3444, 3319, 3173, 2964, 2185, 1655, 1587, 1408 cm−1; 1H NMR (400 MHz, DMSO-d 6 ): δ 5.37 (1H, s, CH), 7.08–7.14 (3H, m, NH2, Ar–H), 7.18–7.22 (1H, m, Ar–H), 7.33 (1H, d, J = 8.04 Hz, Ar–H), 7.38 (2H, d, J = 8.99 Hz, Ar–H), 7.53–7.55 (1H, m, Ar–H), 7.75 (1H, d, J = 9.1 Hz, Ar–H), 7.91 (1H, d, J = 9.04 Hz Ar–H), 8.16 (1H, d, J = 1.51 Hz, Ar–H) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ 37.38, 57.25, 115.29, 118.36, 120.17, 122.03, 125.80, 126.07, 128.73, 129.09, 129.53, 129.74, 130.09, 130.35, 131.07, 132.15, 147.20, 148.10, 159.70, 159.74 ppm; Anal. Calc. for C20H12Br2N2O (%): C, 52.66; H, 2.65; N, 6.14; found: C, 52.81; H, 2.59; N, 5.98.
Supporting information
Supplementary data associated with this article can be found via the “Supplementary Content” section of this article’s webpage.
References
Y. Gu, Green Chem. 14, 2091–2128 (2012)
K. Tanaka, F. Toda, Chem. Rev. 100, 1025–1074 (2000)
I. Ugi, Pure Appl. Chem. 73, 187–191 (2001)
R.A. Sheldon, Green Chem. 7, 267–278 (2005)
P. Anastas, N. Eghbali, Chem. Soc. Rev. 39, 301–312 (2010)
T. Welton, Green Chem. 13, 225 (2011)
D. Chaturvedi, Curr. Org. Chem. 15, 1236–1248 (2011)
C. Yue, D. Fang, L. Liu, T.F. Yi, J. Mol. Liq. 163, 99–121 (2011)
R. Giernoth, Angew. Chem. Int. Ed. 49, 2834–2839 (2010)
H. Yufeng, X. Peng, Effect of the Structures of Ionic Liquids on Their Physical Chemical Properties (Springer, Berlin, 2014), pp. 141–174
C.P. Mehnert, R.A. Cook, N.C. Dispenziere, M. Afeworki, J. Am. Chem. Soc. 124, 12932–12933 (2002)
M.B. Gawande, Y. Monga, R. Zboril, R. Sharma, Coord. Chem. Rev. 288, 118–143 (2015)
R. Skoda-Földes, Molecules 19, 8840–8884 (2014)
B. Xin, C. Jia, X. Li, Curr. Org. Chem. 20, 616–628 (2016)
E.J. Jung, B.H. Park, Y.R. Lee, Green Chem. 12, 2003–2011 (2010)
S.M. Wickel, C.A. Citron, J.S. Dickschat, Eur. J. Org. Chem. 2013, 2906–2913 (2013)
S. Banerjee, A. Horn, H. Khatri, G. Sereda, Tetrahedron Lett. 52, 1878–1881 (2011)
D. Kumar, V.B. Reddy, S. Sharad, U. Dube, S. Kapur, Eur. J. Med. Chem. 44, 3805–3809 (2009)
J.L. Wang, D. Liu, Z.J. Zhang, S. Shan, X. Han, S.M. Srinivasula, C.M. Croce, E.S. Alnemri, Z. Huang, Proc. Natl. Acad. Sci. 97, 7124–7129 (2000)
A.D. Patil, A.J. Freyer, D.S. Eggleston, R.C. Haltiwanger, M.F. Bean, P.B. Taylor, M.J. Cranfa, A.L. Breen, H.R. Bartus, R.K. Johnson, R.P. Hertzberg, J.W. Westley, J. Med. Chem. 36, 4131–4138 (1993)
C.S. Konkoy, D.B. Fick, S.X. Cai, N.C. Lan, J.F.W. Keana, Int. Appl. WO 0075123 2000; Chem. Abstr. 134, 29313a (2001)
S.M.O. Costa, T.L.G. Lemos, O.D.L. Pessoa, C. Pessoa, R. Montenegro, R. Braz-Filho, J. Nat. Prod. 64, 792–795 (2001)
D.L. Wood, D. Panda, T.R. Wiernicki, L. Wilson, M.A. Jordan, J.P. Singh, Mol. Pharmacol. 52, 437–444 (1997)
A.A. Hussein, I. Barberena, T.L. Capson, T.A. Kursar, P.D. Coley, P.N. Solis, M.P.J. Gupta, Nat. Prod. 67, 451–453 (2004)
J.J. Hollick, B.T. Golding, I.R. Hardcastle, N. Martin, C. Richardson, L.J.M. Rigoreau, G.C.M. Smith, R.J. Griffin, Bioorg. Med. Chem. Lett. 13, 3083–3086 (2003)
S. Kumar, D. Hernandez, B. Hoa, Y. Lee, J.S. Yang, A. McCurdy, Org. Lett. 10, 3761–3764 (2008)
A.H.F.A. El-Wahab, H.M. Mohamed, A.M. El-Agrody, A.H. Bedair, Eur. J. Chem. 4, 467–483 (2013)
S. Shinde, G. Rashinkar, R. Salunkhe, J. Mol. Liq. 178, 122 (2013)
M. Ghashang, Res. Chem. Intermed. 42, 4191 (2016)
A.R. Moosavi-Zare, M.A. Zolfigol, O. Khaledian, V. Khakyzadeh, M.H. Beyzavi, H.G. Kruger, Chem. Eng. J. 248, 122–127 (2014)
M. Tajbakhsh, M. Kariminasab, H. Alinezhad, R. Hosseinzadeh, P. Rezaee, M. Tajbakhsh, H.J. Gazvini, M.A. Amiri, J. Iran. Chem. Soc. 12, 1405–1414 (2015)
S.J. Kalita, N. Saikia, D.C. Deka, H. Mecadon, Res. Chem. Intermed. 42, 6863–6871 (2016)
D. Kumar, V.B. Reddy, B.G. Mishra, R.K. Rana, M.N. Nadagouda, R.S. Varma, Tetrahedron 63, 3093–3097 (2007)
R. Teimuri-Mofrad, F. Mirzaei, H. Abbasi, K.D. Safa, C. R. Chim. (2016). doi: 10.1016/j.crci.2016.06.011
R. Teimuri-Mofrad, K.D. Safa, K. Rahimpour, R. Ghadari, J. Organomet. Chem. 811, 14–19 (2016)
A. Shahrisa, R. Teimuri-Mofrad, M. Gholamhosseini-Nazari, Synlett 26, 1031–1038 (2015)
A. Shahrisa, R. Teimuri-Mofrad, M. Gholamhosseini-Nazari, Mol. Divers. 19, 87–101 (2014)
R. Teimuri-Mofrad, A. Shahrisa, M. Gholamhosseini-Nazari, N. Arsalani, Res. Chem. Intermed. 42, 3425–3439 (2016)
Y. He, X. Yu, Mater. Lett. 61, 2071–2074 (2007)
Acknowledgements
The authors thank research affairs of the University of Tabriz for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Teimuri-Mofrad, R., Gholamhosseini-Nazari, M., Payami, E. et al. Novel ferrocene-based ionic liquid supported on silica nanoparticles as efficient catalyst for synthesis of naphthopyran derivatives. Res Chem Intermed 43, 7105–7118 (2017). https://doi.org/10.1007/s11164-017-3061-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3061-x