
Abstract
Pechmann condensation reaction of 3-hydroxyphenol and ethyl acetoacetate in the presence of 1,1′‐butylenebispyridinium hydrogen sulfate as an efficient, green, and recyclable catalyst produces 7-hydroxy-4-methylcoumarin in good yield under solvent-free conditions at room temperature. This catalyst has advantages such as the following: good to excellent yields, short reaction times, simplicity in operation, and easy workup procedure.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Coumarins an important class of compounds whose synthesis has been the focus of attention from many organic and medicinal chemists because of the broad spectrum of their biological and pharmaceutical properties. These properties include anticancer [1], antibacterial [2], inhibitory of HIV-1 protease [3], and inhibition of platelet aggregation [4]. Coumarins are also used as intermediates for the synthesis of chromenes, coumarones, fluorocoumarins, and 2-acylresorcinols [5]. Several synthetic procedures have been used for the synthesis of coumarins, such as Knoevenagel condensation [6–8], Pechmann condensation [9], Claisen [10], Reformatsky [11], and Wittig reaction [12]. However, Pechmann reaction is the most common and simplest procedure for the synthesis of various coumarin-based phenols and β-keto esters or unsaturated carboxylic acids under acid catalysis. In recent years, different acid catalysts have been used in Pechmann condensation [13–18]. Most of these methods have severe drawbacks including the use of a large amount of catalysts, for example, sulfuric acid 10–12 equivalents [13], trifluoroacetic acid 3–4 equivalents [14], and phosphorous pentoxide is required in a fivefold excess [15]. Thus, often high temperatures [16, 17], hard reaction conditions [18], low yield of products being hard to purify, poor selectivity, use of toxic or highly expensive reagents [19, 20], and also the disposal of acidic waste leads to environmental pollution. Various ionic liquids are reported for the synthesis of coumarin [21–23]. In addition, ionic liquids are readily recycled and tunable to specific chemical tasks. (Bbpy)(HSO4)2 is a low-cost, readily available acidic material. Recently, (Bbpy)(HSO4)2 has been used as a versatile mediator or an efficient Brønsted acidic ionic liquid catalyst for the one‐pot multi-component synthesis of unsymmetrical polyhydroquinoline derivatives, which were reported for the first time by Ghaffari Khaligh [24]. As part of our continuing interest in the development of new synthetic methodologies [25–29], we wish to report an efficient and green procedure for the synthesis of coumarin derivatives by Pechmann condensation in the presence of (Bbpy)(HSO4)2 as a catalyst under solvent-free conditions at room temperature (Scheme 1). This procedure is better and more simple than the other procedures to synthesize coumarin derivatives in terms of the amount of the loading catalyst, reaction conditions, product yields, and the required temperature.

Synthesis of coumarin derivatives by Pechmann condensation
Experimental
General
Chemicals were either prepared in our laboratories or they were purchased from Aldrich or Fluka chemical companies. The products were isolated and identified by comparing their physical, melting point, and spectral data with those in the literature. NMR spectra were recorded at room temperature on a Bruker AM 250 spectrometer at 250 MHz for 1H and 62.5 MHz for 13C, respectively, using CDCl3 as solvent. Chemical shifts are expressed in δ values (ppm) relative to TMS as internal standard for 1H and relative to TMS (0.00 ppm) or to CDCl3 (77.05 ppm) for 13C NMR spectra. Melting points were determined on an electrical melting point apparatus in an open capillary and uncorrected. The progress of the reaction was followed with thin-layer chromatography (TLC) using silica gel SILG/UV 254 and 365 plates.
Synthesis of 1,1′‐butylenebispyridinium hydrogen sulfate (Bbpy)(HSO4)2
In this study, pyridine (0.95 g, 12.01 mmol), 1,4‐dichlorobutane (0.80 g, 6.30 mmol), and dry acetonitrile (10 mL) were added into a two‐neck 100-mL round bottomed flask equipped with a reflux condenser and magnetic stirrer. The mixture was refluxed for 48 h. After the reaction, the solvent was removed under vacuum, and the residue was washed with dichloromethane and dried at 60 °C under vacuum to give 1,1‐butylenebispyridinium dichloride (Bbpy)Cl2 as a white solid (1.68 g, 98.2 %). To a stirred solution of (Bbpy)Cl2 (1.40 g, 4.91 mmol) in 25 mL dry CH2Cl2 at 0 °C was added dropwise 98 % H2SO4 (0.53 mL, 9.82 mmol) over 10 min. The resulting solution was refluxed for 48 h, and then the solution was washed with a mixture of ethanol and water (50 %, 2 × 5 mL). The solvent was distilled off under reduced pressure to give (Bbpy)(HSO4)2 as a darkish viscous liquid (1.96 g, 98 %) [24].
General procedure for the coumarin derivatives 3a–3m
In a round-bottom flask (25 mL), (Bbpy)(HSO4)2 (10 mg, 2.4 mol %) was added to the mixture of 3-hydroxy phenol (5.0 mmol) and ethyl acetoacetate (5.0 mmol) and the reaction mixture stirred at ambient temperature for the appropriate time (Table 2). The progress of reaction was monitored by TLC (eluent, n-hexane:ethyl acetate, 4:1). After the completion of the reaction, water (10.0 mL) was added to the reaction mixture, and the catalyst was recovered by filtration. Evaporation of the solvent from the filtrate and recrystallization of the solid residue from hot ethanol afforded the pure products in good to excellent yields. All compounds were identified by comparison of melting point, 1H NMR, and 13C NMR with those reported. Spectroscopic data for selected examples listed below.
7-Amino-4-methyl-2H-chromen-2-one (Table 2, Entry 4)
1H-NMR (250 MHz; CDCl3): δ 7.35–7.39 (d, J = 8.5 Hz, 1H), 6.52–6.56 (dd, J = 6 Hz, J = 2.25 Hz, 1H), 6.37–6.38 (d, J = 2.25 Hz, 1H), 6.06 (broad s, 2H), 5.873-5.877 (s, 1H), 2.272-2.276 (s, 3H); 13C-NMR (62.9 MHz, CDCl3): δ 161.2, 155.9, 154.2, 153.5, 126.6, 111.6, 109.3, 107.9, 98.9, 18.4.
7,8-Dihydroxy-4-methyl-2H-chromen-2-one (Table 2, Entry 5)
1H-NMR (250 MHz; CDCl3): δ 9.12–10.15 (broad s, 2H), 7.03–7.07 (d, J = 8.5 Hz, 1H), 6.69–6.80 (d, J = 8.5 Hz, 1H), 6.08 (s, 1H), 2.31 (s, 3H) ppm. 13C-NMR (62.9 MHz, CDCl3): δ 160.6, 154.3, 149.8, 143.8, 132.6, 115.9, 113.2, 112.6, 110.6, 18.7.
4-Methyl-2H-benzo[h]chromen-2-one (Table 2, Entry 10)
1H-NMR (250 MHz; CDCl3): δ 8.32–8.35 (m, 1H), 8.00-8.03 (m, 1H), 7.65–7.86 (m, 4H), 6.480 (s, 1H), 2.506 (s, 3H); 13C-NMR (62.9 MHz, CDCl3): δ 160.1, 154.6, 134.8, 129.1, 128.4, 127.8, 124.4, 122.6, 122.0, 121.7, 115.5, 114.3, 19.1.
6-Hydroxy-4-methyl-2H-chromen-2-one (Table 2, Entry 13)
1H-NMR (250 MHz; CDCl3): δ 9.71 (s, 1H), 7.18–7.22 (broad s, 1H), 7.00 (broad s, 2H), 6.332 (s, 1H), 2.342 (s, 3H), 13C-NMR (62.9 MHz, CDCl3): δ 160.5, 154.2, 153.2, 146.7, 120.6, 120.2, 117.8, 114.9, 110.0, 18.5.
Results and discussion
1,1′‐butylenebispyridinium hydrogen sulfate (Bbpy)(HSO4)2 was prepared readily in the two‐step process as detailed in the literature (Scheme 2). In first step, two equivalents of pyridinium was alkylated with 1,4‐dichlorobutane in acetonitrile under reflux conditions for 24 h. In the second step, by adding H2SO4 in dry CH2Cl2 under reflux conditions for 48 h the resulting halide 1,1′‐butylenebispyridinium dichloride (Bbpy)Cl2 converted to 1,1′‐butylenebispyridinium hydrogen sulfate (Bbpy)(HSO4)2 and was isolated as a viscous liquid.

Synthesis of 1,1′‐butylenebispyridinium hydrogen sulfate (Bbpy)(HSO4)2
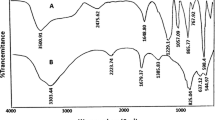
An informative technique for an investigation of catalyst formation is FT-IR spectroscopy. The FT-IR spectrum of the catalyst (Fig. 1) was recorded using the KBr disc technique. The O=S=O asymmetric and symmetric stretching modes of the sulfonic acid functional groups were found in the ranges 1020–1060 cm−1, respectively, and that of the S–O stretching mode at 1227 cm−1. Also, one absorption was seen at 1637 for C=N cm−1 and 1400 cm−1 for the C=C group. The spectrum also showed a broad OH stretching absorption around 3144 and 3465 cm−1.

The FT-IR spectrum of 1,1‐butylenebispyridinium hydrogen sulfate (KBr disc)
For understanding the efficiency of 1,1‐butylenebispyridinium hydrogen sulfate to the Pechmann condensation, the reaction of ethyl acetoacetate 1 (5 mmol) with resorcinol 2 (5 mmol) under solvent-free conditions at room temperature was selected as the model reaction (Scheme 1). Firstly, when the reaction was carried out in the absence of 1,1‐butylenebispyridinium hydrogen sulfate, the reaction was slow and 7-hydroxy-4-methylcoumarin weren’t formed after 24 h (Table 1, Entry 1). The results listed in Table 1 show that when the reaction was performed in the presence of 3 mg of 1,1‐butylenebispyridinium hydrogen sulfate, coumarin 3a was achieved in good yield (Table 1, Entry 3). The best result was obtained when we used 10 mg of 1,1‐butylenebispyridinium hydrogen sulfate, which leads to the formation of the 7-hydroxy-4-methylcoumarin in excellent yield and with short reaction times (95 %, 25 min) (Table 1, Entry 5). When the reaction was carried out in 15 mg of 1,1‐butylenebispyridinium hydrogen sulfate, there was less yield of product (Table 1, Entry 6). Increasing the amount of catalyst until 15 mg had no positive effect on the product yield (Table 1, Entries 7–9). After optimization of the reaction conditions, other phenols were examined with ethyl acetoacetate in the presence of 10 mg of 1,1‐butylenebispyridinium hydrogen sulfate under solvent-free conditions and at room temperature. The results are presented in Table 2.
The results listed in Table 2 show that the rate of reactions were increased in the presence resorciniol (Table 2, Entry 1), pyrogallol (Table 2, Entry 5), phloroglucinol (Table 2, Entry 6), orcinol (Table 2, Entry 7), 2,6-dihydroxy toluene (Table 2, Entry 8), 4-nitro phenol (Table 2, Entry 12), and hydroquinone (Table 2, Entry 13) and produce coumarin derivatives in excellent yields. Also, 3-aminophenol (Table 2, Entry 4) reacted to provide the amino coumarin derivatives in good yield with slightly longer reaction times. 1-Naphtol (Table 2, Entry 10) reacted with longer reaction time and low yield due to having another phenyl group. Similarly, phenol (Table 2, Entry 9) reacted with longer reaction time and very low yield, but on the other hand, formation of 4-methylcoumarin in acceptable yield from the reaction of unsubstituted phenol. The products obtained were purified by recrystallisation in hot ethanol, and the structures of the products 3a–3m were deduced from the 1H NMR and 13C NMR spectra. For example, the 1H NMR spectrum of 3e exhibited one singlet for OH group at 6.08 ppm, one singlet for CH3 protons at 2.31 ppm, two doublets at 7.03–7.07 (d, 3 J = 8.5 Hz), and 6.76–6.80 (d, 3 J = 8.5 Hz) for C–H proton. The 13C NMR spectrum of 3e displayed ten distinct resonances in agreement with the proposed structure of product. The reaction pathway proposed for the Pechmann condensation of resorcinol with ethyl acetoacetate in the presence of (Bbpy)(HSO4)2 as the catalyst has been described (Scheme 3). It is suggested that ethyl acetoacetate in the presence of catalyst produce intermediate 4. Resorcinol react with 4 and generate intermediate A.

Proposed mechanism for the Pechmann condensation of resorcinol with ethyl acetoacetate in the presence of (Bbpy)(HSO4)2 catalyst
Interamolecular cyclization and elimination EtOH led to formation intermediate B. Finally, the intermediate B undergoes elimination of water to give target product 3a.
In order to show the merit of (Bbpy)(HSO4)2 with recently reported protocols, some homogeneous and heterogeneous catalysts in the Pechmann condensation with resorciniol (1) and ethyl acetoacetate (2) are presented in Table 3. The results clearly show that some catalysts such CMK-5-SO3H (Table 3, Entry 1), iodine (Table 3, Entry 2), chloroaluminate (Table 3, Entry 9), Mont-K10 (Table 3, Entry 10), and Mont-KSF (Table 3, Entry 11) requires high temperatures and longer reaction time for completion of the reaction. On the other hand, polystyrene-supported GaCl3 and PVPP-BF3 (Table 3, Entry 5) used in reflux conditions, which are hard reaction conditions. Also, some used from toxic solvents such as toluene, which had dangerous reaction conditions and much longer reaction times (Table 3, Entry 2). As can be seen, our procedure was simpler and more efficient, without the need for temperature and toxic solvents, and used a lower amount of the catalyst, which shows (Bbpy)(HSO4)2 is a powerful catalyst for the synthesis coumarin derivatives, thus without doubt, our procedure is a clean and environmentally friendly reaction.
The recoverability and reusability of (Bbpy)(HSO4)2 was checked for the synthesis of coumarin derivatives. It was found that the (Bbpy)(HSO4)2 could be reused several times without any appreciable loss of activity (Table 4). After the completion of the reaction of ethyl acetoacetate (5 mmol), resorcinol (5 mmol), and (Bbpy)(HSO4)2 (10 mg) as a model reaction, the catalyst was filtered, washed with chloroform (10 mL). As depicted in Table 4, the recoverability and reusability of catalyst was also checked for the synthesis of other coumarin derivatives. This process was repeated for five cycles, and each run afforded the desired product in good to excellent yield. The decrease in activity can be mainly attributed to unavoidable loss of the catalyst during the process of collection and washing.
Conclusion
In summary, an efficient and environmentally friendly procedure for the synthesis of coumarin derivatives using (Bbpy)(HSO4)2-catalyzed through Pechmann condensation reaction between ethyl acetoacetate with various phenol under solvent-free conditions and at ambient temperature was described. Mild reaction conditions, short reaction times, good to excellent yield independent of temperature, simple experimental and product isolation procedures are the significant advantages of the present method.
References
C.J. Wang, Y.J. Hsieh, C.Y. Chu, Y.L. Lin, T.H. Tseng, Cancer Lett. 183, 163 (2002)
O. Kayser, H. Kolodziej, Planta Med. 63, 508 (1997)
S. Kirkiacharian, D.T. Thuy, S. Sicsic, R. Bakhchinian, R. Kurkjian, T. Tonnaire, Il Farmaco 57, 703 (2002)
G. Cavettos, G.M. Nano, G. Palmisano, S. Tagliapietra, Tetrahedron 12, 707 (2001)
S.M. Sethna, N.M. Shah, Chem. Rev. 36, 1 (1945)
A. Shaabani, R. Ghadari, A. Rahmati, A.H. Rezayan, J. Iran. Chem. Soc. 6, 710 (2009)
G. Jones, Org. React. 15, 204 (1967)
G. Brufola, F. Fringuelli, O. Piermatti, F. Pizzo, Heterocycles 43, 1257 (1996)
H. Pechmann, Chem. Ber. 17, 929 (1884)
N. Cairns, L.M. Harwood, D.P. Astles, Chem. Soc. Perkin Trans. 1, 3101 (1994)
I. Yavari, R. Hekmat Shoar, A. Zonouzi, Tetrahedron Lett. 39, 2391 (1998)
J.R. Johnson, Org. React. 1, 210 (1942)
A.D. Patil, A.J. Freyer, S.E. Drake, R.C. Haltiwanger, M.F. Bean, P.B. Taylor, M.J. Caranfa, A.L. Breen, H.R. Bartus, R.K. Johnson, R.P. Hertzberg, J.W. Westley, J. Med. Chem. 36, 4131 (1993)
L.L. Woods, J. Sapp, J. Org. Chem. 27, 3703 (1962)
A. Robertson, W.F. Sandrock, C.B. Henry, J. Chem. Soc. 1931, 2426 (1931)
D. Zareyee, M. Serehneh, J. Mol. Catal. A Chem. 391, 88 (2014)
B. Karimi, D. Zareyee, Org. Lett. 10, 3989 (2008)
B. Rajithaa, V.N. Kumar, P. Someshwar, J.V. Madhava, P.N. Reddy, Y.T. Reddy, Arkivoc Xii, 23 (2006)
J. DeGrote, S. Tyndall, K.F. Wong, M. VanAlstine-Parris, Tetrahedron Lett. 55, 6715 (2014)
D. Prajapati, M. Gohain, Catal. Lett. 119, 59 (2007)
M.K. Potdur, S.S. Mohile, M.M. Salunkhe, Tetrahedron Lett. 42, 9285 (2001)
A.C. Kandekar, B.M. Khadikar, Synlett 1, 152 (2002)
M.K. Potdar, M.S. Rasalkar, S.S. Mohile, M.M. Salunkhe, J. Mol. Catal. A Chem. 235, 249 (2005)
N.G. Khaligh, Chin. J. Catal. 35, 1497 (2014)
S. Sajjadifar, S. Rezayati, Chem. Pap. 68, 531 (2014)
S. Rezayati, Z. Erfani, R. Hajinasiri, Chem. Pap. 69, 536 (2015)
S. Rezayati, S. Sajjadifar, J. Sci. I. R. Iran 25, 329 (2014)
R. Hajinasiri, S. Rezayati, Zeitschrift für Naturforschung B (Z. Naturforsch.) 68b, 818 (2013)
S. Rezayati, S. Sajjadifar, R. Hajinasiri, Iran J Sci Technol 39A2, 179 (2015)
A. Rahmatpour, S. Mohammadian, C. R. Chim. 16, 271 (2013)
S.S. Bahekar, D.B. Shinde, Tetrahedron Lett. 45, 7999 (2004)
P.G. Mandhane, R.S. Joshi, A.R. Ghawalkar, G.R. Jadhav, G.H. Gill, Bull. Korean Chem. Soc. 30, 2969 (2009)
P.K. Sahua, P.K. Sahub, D.D. Agarwal, J. Mol. Catal. A Chem. 395, 251 (2014)
J.C. Rodriguez-Dominguez, G. Kirsch, Tetrahedron Lett. 47, 32799 (2006)
S.V. Atghia, S.S. Beigbaghlou, C. R. Chim. 17, 1155 (2014)
G.P. Romanelli, D. Bennardi, D.M. Ruiz, G. Baronetti, H.J. Thomas, J.C. Autino, Tetrahedron Lett. 45, 8935 (2004)
T. Li, Z. Zhang, F. Yang, J. Chem. Res. 1998, 38 (1998)
B. Karami, M. Kiani, M.A. Hoseini, Chin. J. Catal. 35, 1206 (2014)
R.S. Keri, K.M. Hosamani, H.R.S. Reddy, Catal. Lett. 131, 321 (2009)
Acknowledgments
We gratefully acknowledge financial and spiritual support from Islamic Azad University of Qaemshahr and Gonbad Kavous University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rezayati, S., Sheikholeslami-Farahani, F., Rostami-Charati, F. et al. One-pot synthesis of coumarine derivatives using butylenebispyridinium hydrogen sulfate as novel ionic liquid catalyst. Res Chem Intermed 42, 4097–4107 (2016). https://doi.org/10.1007/s11164-015-2261-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-015-2261-5