
Abstract
An expeditious method with microwave irradiation has been developed for synthesis of novel Schiff bases from dimers of 4-amino-3-[3-(1-benzyl)indole]-5-thiomethyl-1,2,4-triazole. Its distinct advantages are short reaction times and good conversion. The structures of these new Schiff bases were established by 1H NMR, IR, and mass spectroscopy, and elemental analysis. The antibacterial activity of ten novel Schiff bases against three bacterial strains was studied by the disk diffusion method. Preliminary results indicated that some of the compounds had strong antibacterial activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the last few years derivatives of indole have attracted much attention because of their special biological activity in medicine and agriculture. Andreani et al. [1–6] reported that indole derivatives have substantial biological activity, for example cytotoxic, antibacterial, antiviral, anticancer, and antiproliferative, and as plant growth regulators. Furthermore, the triazole structure is occurs widely in natural medicines and synthetic drugs. Currently available anti-microbial [7] or anti-tumor [8] drugs, for example terconazole, itraconazole, fluconazole, fosfluconazole, vibunazole, letrozole, anastrozole, and vorozole all contain the triazole ring. Moreover, Schiff base compounds [9–13] and heterocyclic sulfide compounds [14] were also reported to have cytotoxic, anticonvulsant, antiproliferative, anticancer, antifungal, and antibacterial activity. In view of these research results and because of the principle of active superposition, we synthesized a series of triazole Schiff bases containing indole and heterocyclic sulfide structures. We can expected this kind of new triazole Schiff base to have good biological activity. We also hoped they could be used for other applications in the chemical industry.
Microwave synthesis, because of its unique advantages, has been widely applied in organic synthesis in recent years [15–17]. We are interested in the pharmaceutical synthesis using microwave methods [18–21]. As a continuation of this work, herein we report the synthesis, under microwave irradiation, of novel triazole Schiff bases containing indole and heterocyclic sulfide structures. The synthetic route is shown in Scheme 1.

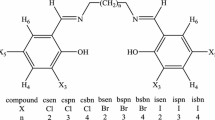
Synthetic route for new Schiff bases derived from dimers of 4-amino-3-[3-(1-benzyl)indole]-5-thiomethyl-1,2,4-triazole
Results and discussion
Spectroscopic studies
The structures of 6 and 7a–j were confirmed on the basis of spectral data and elemental analysis. The IR spectrum of 6 contained an absorption band at 3,326–3,123 cm−1, because of the NH2 group; this was absent from the IR spectra of 7a–j. Strong bands at 1,588–1,526 cm−1 were assigned to absorption of C=N. The 1H NMR spectrum of 6 contained a singlet peak at 6.16 ppm because of the NH2 group; this was absent from the spectra of compounds 7a–j. The singlet peak at 9.10–8.45 ppm was assigned to ArCHN. The protons of ArH appeared at 8.69–6.91 ppm. The singlet peak at approximately 5.45 ppm was assigned to ArCH2N. The singlet peak at approximately 3.53 ppm was assigned to SCH2CH2S. In the 13C NMR spectra of compounds 6 and 7a–j, the peak at approximately 50 ppm was assigned to PhCH2N, the carbon atom of SCH2CH2S appeared at approximately 33 ppm. Other peaks were assigned to carbon atoms of the aromatic nucleus and the indole ring. Their ESI mass spectra contained the correct molecular ions with high intensity.
Antibacterial studies
The newly synthesized compounds were screened for their antibacterial activity against Escherichia coli (ATCC-25922), Staphylococcus aureus (ATCC-25923), and Pseudomonas aeruginosa (ATCC-27853) bacterial strains by the disk-diffusion method [22, 23]. A standard inoculum, prepared from beef extract, peptone, sodium chloride, blood serum, and agar, was applied to the surface of sterile agar plates. A sterile glass spreader was used for even distribution of the inoculum. Disks 6.25 mm in diameter were prepared from ordinary filter paper and sterilized by dry heat at 140 °C for an hour. The sterile disks, previously soaked in a fixed concentration (500 μg/mL) of the test compounds, were placed on the nutrient agar medium. The plates were inverted and incubated at 37 °C for 24 h. Solvent and growth control experiments were also conducted. Sparfloxacin was used as standard drug. The results from testing of bacterial inhibition are list in Table 1.
As is obvious from Table 1, among the compounds screened, compounds 7b, 7d, and 7f are the most biologically effective. Compound 7f had good antibacterial activity against all three bacteria, compound 7b had satisfactory activity against E. coli and P. aeruginosa, and compound 7d strongly inhibited growth of S. aureus. So, we have reason to believe these three compounds or their derivatives can be developed as antibacterial drugs.
Comparison of microwave irradiation and conventional heating
As shown in Table 2, microwave irradiation had two large advantages over conventional heating—reaction times were reduced from 1,440–1,800 min to 4–8 min, i.e. reaction was 225–360 times faster, and yields increased from 72–78 % to 85–94 %. We can therefore conclude that the microwave-enhanced procedure enables rapid and efficient synthesis of triazole Schiff base compounds containing indole and heterocyclic sulfide structures.
Experimental
Melting points were measured on a micro-melting point apparatus and are uncorrected. Infrared spectra were obtained with a 1700 Perkin–Elmer FTIR using KBr disks. NMR spectra were recorded on a Varian Inova 400-MHz spectrometer with TMS as internal standard, DMSO-d 6 or pyridine-d 5 as solvent, operating at 400 MHz for 1H and 100 MHz for 13C. Mass spectra were determined on Finnigan LCQDECA instrument. Elemental analysis was performed with a Carlo Erba 1106 autoanalyzer. All microwave-assisted reactions were performed in a commercial microwave reactor (XH-100A, 100–1,000 W; Beijing Xianghu Science and Technology Development, Beijing, PR China). All solvents were purified before use. Intermediates 2, 3, 4, and 5 were prepared in accordance with reported procedures [24, 25].
Procedure for preparation of intermediate 6
Compound 5 (1.7 mmol), KOH (1.6 mmol), 1,2-dibromoethane (0.8 mmol), and methanol (5 mL) were placed in a vessel which was then sealed and shaken to homogenize the mixture. The mixture was irradiated at 350 W for 10 min. After cooling to room temperature, filtration, and recrystallization, we obtained the pure compound 6 as a white solid, yield 91 %, m.p. 247–249 °C; IR (KBr) (cm−1): 3326, 3123, 1622, 1581, 1448, 1386, 1338, 1187, 1021, 930, 740; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.50 (s, 2H, ArH), 8.31(d, J = 7.6 Hz, 2H, ArH), 7.55(d, J = 8.0 Hz, 2H, ArH), 7.33–7.15 (m, 14H, ArH and indole-CH); 6.16(s, 4H, NH2), 5.46 (s, 4H, PhCH2N), 3.34 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, DMSO-d 6 , ppm) δ: 152.08, 151.10, 138.14, 136.06, 129.71, 129.06, 127.98, 127.53, 126.45, 122.90, 122.24, 121.03, 111.00, 102.08, 49.91, 31.91; ESI–MS m/z (%): 669 [(M + 1)+, 100]. Anal. Calcd. for C36H32N10S2: C, 64.65; H, 4.82, N, 20.94. Found: C, 64.70; H, 4.75; N, 20.87 %.
Conventional method for preparation of compounds 7a–j
Compound 6 (0.12 mmol), aldehyde (0.27 mmol), and glacial acetic acid (5 mL) were placed in a round-bottomed flask and the reaction mixture was heated to reflux for 24–30 h. When the reaction was complete (monitored by TLC) the flask was cooled to room temperature and evaporated to remove the glacial acetic acid. Ethanol (5 ml) was added into the residue. Solid formed immediately and was collected by filtration and recrystallized from N,N-dimethylformamide to furnish the pure compounds 7a–j.
Microwave method for preparation of compounds 7a–j
Compound 6 (0.12 mmol), acetic acid (2 mL), and aldehyde (0.27 mmol) were mixed thoroughly in a sealed vessel. The vessel was then placed in a microwave oven and the mixture was irradiated at 300 W for 4–8 min. The reaction was monitored by TLC until it was complete. The acetic acid was then removed by reduced-pressure distillation. Ethanol (10 mL) was added to the residue. The solid formed was isolated by filtration. After recrystallization, the pure product 7a–j was obtained as a white, yellow, or green solid. The physical and spectra data of compounds 7a–j are as follows.
7a: Yellow solid, yield 86 %, m.p. 230–232 °C; IR (KBr)(cm−1): 3055, 1578, 1439, 1387, 747, 693; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.88 (s, 2H, ArCH=N), 8.27 (d, J = 7.6 Hz, 2H, ArH), 7.90–7.87 (m, 6H, ArH), 7.66–7.52 (m, 8H, ArH), 7.26–7.16 (m, 14H, ArH and indole-CH), 5.44 (s, 4H, PhCH2N), 3.53 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 165.48, 144.49, 137.54, 137.02, 136.12, 135.09, 133.31, 132.54, 129.96, 129.70, 129.51, 129.24, 128.23, 127.78, 127.23, 123.06, 121.71, 110.80, 103.20, 50.26, 33.80; ESI–MS m/z (%): 845 [(M + 1)+, 100]. Anal. Calcd. for C50H40N10S2: C, 71.07; H, 4.77, N, 16.58. Found: C, 71.13; H, 4.70; N, 16.54 %.
7b: Pale yellow solid, yield 85 %, m.p. 232–234 °C; IR (KBr)(cm−1): 3063, 1615, 1583, 1449, 1379, 1270, 742; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.91 (s, 2H, ArCH=N), 8.25 (d, 2H, ArH), 7.90 (s, 2H, ArH), 7.74–7.50 (m, 10H, ArH), 7.26–7.15 (m, 14H, ArH and indole-CH), 5.44 (s, 4H, PhCH2N), 3.53 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 164.42, 163.97, 161.97, 144.44, 137.38, 136.88, 134.62, 131.50, 131.42, 129.89, 129.10, 128.13, 127.69, 127.14, 127.02, 125.88, 122.96, 121.60, 120.13, 119.91, 115.24, 115.01, 110.77, 102.85, 50.20, 33.81; ESI–MS m/z (%): 881 [(M + 1)+, 100]. Anal. Calcd. for C50H38F2N10S2: C, 68.16; H, 4.35; N, 15.90. Found: C, 68.21; H, 4.29; N, 15.87 %.
7c: Pale yellow solid, yield 87 %, m.p. 209–211 °C; IR (KBr)(cm−1): 3242, 3023, 1587, 1442, 1371, 1164, 737, 693; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 10.56 (s, 2H, ArOH), 9.00 (s, 2H, ArCH=N), 8.28 (d, J = 7.6 Hz, 2H, ArH), 7.96(s, 2H, ArH), 7.88(d, J = 7.6 Hz, 2H, ArH), 7.59 (d, J = 8.0 Hz, 2H, ArH), 7.46 (t, J = 8.4 Hz, 2H, ArH), 7.24–7.16 (m, 14H, ArH and indole-CH), 7.00–6.92 (m, 4H, ArH), 5.46 (s, 4H, PhCH2N), 3.56 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 162.13, 158.91, 143.22, 136.08, 135.47, 133.60, 128.48, 127.73, 127.70, 126.68, 126.22, 125.82, 125.82, 125.80, 121.98, 121.73, 120.14, 118.80, 117.86,116.06, 109.38, 101.83, 48.95, 32.36; ESI–MS m/z (%): 877 [(M + 1)+, 100]. Anal. Calcd. for C50H40N10O2S2: C, 68.47; H, 4.60; N, 15.97. Found: C, 68.53; H, 4.53; N, 15.95 %.
7d: White solid, yield 86 %, m.p. 210–212 °C; IR (KBr)(cm−1): 3038, 1588, 1427, 1249, 1161, 1023, 752; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.71 (s, 2H, ArCH=N), 8.28 (d, J = 7.6 Hz, 2H, ArH), 7.85 (t, J = 8.8 Hz, 6H, ArH), 7.59 (d, J = 8.0 Hz, 2H, ArH), 7.24–7.18 (m, 14H, ArH and indole-CH), 7.08 (d, J = 8.4 Hz, 4H, ArH), 5.43 (s, 4H, PhCH2N), 3.85 (s, 6H, OCH3), 3.51 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 171.43, 164.38, 147.92, 136.09, 135.50, 130.07, 128.38, 127.73, 126.67, 126.19, 125.74, 123.57, 122.58, 121.85, 121.85, 121.55, 120.16, 113.77, 109.37, 101.85, 54.16, 48.87, 32.40; ESI–MS m/z (%): 905 [(M + 1)+, 100]. Anal. Calcd. for C52H44N10O2S2: C, 69.00; H, 4.90; N, 15.48. Found: C, 69.09; H, 4.84; N, 15.45 %.
7e: White solid, yield 91 %, m.p. 269–271 °C; IR (KBr)(cm−1): 3233, 3023, 1587, 1442, 1371, 1265, 1164, 737; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 10.50 (s, 2H, PhOH), 8.70 (s, 2H, ArCH=N), 8.27 (d, J=7.6 Hz, 2H, ArH), 7.85–7.74 (m, 6H, ArH), 7.57 (d, J = 8.0 Hz, 2H, ArH), 7.26–7.16 (m, 14H, ArH and indole-CH), 6.91 (d, J = 8.4 Hz, 4H, ArH), 5.46 (s, 4H, PhCH2N), 3.32 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 165.05, 162.55, 148.19, 143.18, 136.08, 135.47, 130.58, 128.35, 127.70, 126.64, 126.20, 125.77, 122.03, 121.86, 121.77, 120.12, 115.80, 109.34, 101.92, 48.88, 32.37; ESI–MS m/z (%): 877[(M + 1)+, 100]. Anal. Calcd. for C50H40N10O2S2: C, 68.47; H, 4.60; N, 15.97. Found: C, 68.55; H, 4.58; N,15.94 %.
7f: Pale white solid, yield 86 %, m.p. 216–218 °C; IR (KBr)(cm−1): 3054, 2924, 1585, 1437, 1372, 1168, 740; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.89 (s, 2H, ArCH=N), 8.24 (d, J = 8.0 Hz, 2H, ArH), 8.09 (s, 2H, ArH), 7.89–7.82 (m, 6H, ArH), 7.60–7.46 (m, 4H, ArH), 7.29–7.15 (m, 14H, ArH), 5.43 (s, 4H, PhCH2N), 3.53 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 161.88, 143.06, 136.06, 135.51, 133.20, 130.45, 129.89, 128.55, 127.77, 127.70, 126.76, 126.66, 126.30, 125.78, 125.67, 121.62, 120.24, 109.40, 101.51, 48.87, 32.45; ESI–MS m/z (%): 1003 [(M + 1)+, 100]. Anal. Calcd. for C50H38Br2N10S2: C, 59.88; H, 3.82; N,13.97. Found: C, 59.91; H,3.76; N, 13.95 %.
7g: Yellow solid, yield 90 %, m.p. 231–233 °C; IR (KBr)(cm−1): 3071, 2365, 1578, 1526, 1439, 1347, 738, 688; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 9.09 (s, 2H, ArCH=N), 8.69 (s, 2H, ArH), 8.43 (d, J = 8.4 Hz, 2H, ArH), 8.31–8.23 (m, 4H, ArH), 7.93 (s, 2H, ArH), 7.80 (t, J = 8.0 Hz, 2H, ArH), 7.59 (d, J = 8.0 Hz, 2H, ArH), 7.21–7.15 (m, 14H, ArH and indole-CH), 5.44 (s, 4H, PhCH2N), 3.56 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 161.34, 144.62, 138.10, 137.63, 135.07, 134.47, 133.70, 130.87, 130.23, 130.11, 129.28, 128.26, 128.06, 127.79, 127.29, 127.14, 126.89, 124.60, 123.02, 121.83, 116.85, 50.11, 32.33; ESI–MS m/z (%): 935 [(M + 1)+, 100]. Anal. Calcd. for C50H38N12O4S2: C, 64.23; H, 4.10; N, 17.98. Found: C, 64.29; H, 4.02; N, 17.96 %.
7h: Pale green solid, yield 87 %, m.p. 235–237 °C; IR (KBr)(cm−1): 3036, 1576, 1423, 1390, 1299, 1005, 750; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.87 (s, 2H, ArCH=N), 8.25 (d, J = 8.0 Hz, 2H, ArH), 7.87 (s, 2H, ArH), 7.81–7.71 (m, 8H, ArH), 7.58 (d, J = 8.4 Hz, 2H, ArH), 7.26–7.16 (m, 14H, ArH and indole-CH), 5.48 (s, 4H, PhCH2N), 3.52 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, DMSO-d 6 , ppm) δ: 166.42, 148.52, 143.56, 137.10, 135.68, 132.27, 130.67, 128.46, 127.48, 127.26, 126.95, 125.53, 122.58, 121.41, 120.74, 115.80, 110.62, 100.95, 49.18, 29.46; ESI–MS m/z (%): 1003 [(M + 1)+, 100]. Anal. Calcd. for C50H38Br2N10S2: C, 59.88; H, 3.82; N 13.97. Found: C 59.92, H 3.73, N 13.94 %.
7i: Pale white solid, yield 94 %, m.p. 240–242 °C; IR (KBr)(cm−1): 3058, 2925, 1580, 1442, 1419, 1202, 1169, 1026, 745; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 9.10 (s, 2H, ArCH=N), 8.18(d, J = 8.4 Hz, 2H, ArH), 8.03–8.00 (m, 4H, ArH), 7.80 (t, J = 8.0 Hz, 2H, ArH), 7.61–7.47 (m, 6H, ArH), 7.28–7.15 (m, 14H, ArH and indole-CH), 5.49 (s, 4H, PhCH2N), 3.60 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 160.13, 143.28, 136.55, 136.17, 135.61, 133.56, 132.74, 132.58, 130.16, 128.80, 128.04, 127.09, 126.72, 126.03, 125.78, 125.63, 120.20, 109.44, 101.58, 48.84, 32.41; ESI–MS m/z (%): 1003 [(M + 1)+, 100]. Anal. Calcd. for C50H38Br2N10S2: C, 59.88; H, 3.82; N,13.97. found: C, 59.94; H, 3.78; N; 13.95 %.
7j: Yellow solid, yield 88 %, m.p. 214–216 °C; IR (KBr)(cm−1): 3031, 1583, 1442, 1386, 1291, 1156, 736, 702; 1H NMR (400 MHz, DMSO-d 6 , ppm) δ: 8.76 (s, 2H, ArCH=N), 8.25 (d, J = 7.6 Hz, 2H, ArH), 8.11 (s, 2H, ArH,), 7.91 (s, 2H, ArH), 7.57 (d, J = 8.0 Hz, 2H, 5-furyl-H), 7.34 (d, J = 3.6 Hz, 2H, 3-furyl-H), 7.29–7.16 (m, 14H, ArH), 6.80–6.79 (m, 2H, 4-furyl-H), 5.47 (s, 4H, PhCH2N), 3.52 (s, 4H, SCH2CH2S); 13C NMR (100 MHz, Pyridine-d 5 , ppm) δ: 153.34, 148.40, 148.26, 144.64, 137.60, 136.95, 130.24, 129.52, 129.22, 128.15, 127.64, 127.29, 123.06, 121.69, 120.46, 113.45, 110.91, 103.06, 50.46, 33.95; ESI–MS m/z (%): 825 [(M + 1)+, 100]. Anal. Calcd. for C46H36N10O2S2: C, 66.97; H, 4.40; N, 16.98. Found: C, 67.90; H, 4.38; N,16.94 %.
Conclusions
We have developed a facile and efficient method for synthesis, under microwave irradiation, of new Schiff bases derived from dimers of 4-amino-3-[3-(1-benzyl)indole]-5-thiomethyl-1,2,4-triazole. The reactions proceeded much more quickly and yields were higher than when conventional heating was used. Many of the newly synthesized compounds had good antibacterial activity, especially those containing methoxyphenyl and halogen substituents. We therefore believe there is ample scope for further study and our work may have a beneficial outcome.
References
R. Martínez, A.C. Sosa, M.T.R. Apan, Bioorg. Med. Chem. 15, 3912–3918 (2007)
N.A. Meanwell, O.B. Wallace, H. Wang, M. Deshpande, B.C. Pearce, A. Trehan, K.S. Yeung, Z.L. Qiu, J.J.K. Wright, B.A. Robinson, Y.F. Gong, H.G.H. Wang, W.S. Blair, P.Y. Shi, P.F. Lin, Bioorg. Med. Chem. Lett. 19, 5136–5139 (2009)
S.T. Nguyen, M.M. Butler, L. Varady, N.P. Peet, T.L. Bowlin, Bioorg. Med. Chem. Lett. 20, 5739–5742 (2010)
V. Virsodia, A. Manvar, K. Upadhyay, R. Loriya, D. Karia, M. Jaggi, A. Singh, R. Mukherjee, M.S. Shaikh, E.C. Coutinho, A. Shah, Eur. J. Med. Chem. 44, 1355–1362 (2009)
S. Libnow, K. Methling, M. Hein, D. Michalik, M. Harms, K. Wende, A. Flemming, M. Köckerling, H. Reinke, P.J. Bednarski, M. Lalk, P. Langer, Bioorg. Med. Chem. 16, 5570–5783 (2008)
V. Ambrogi, F. Famiani, L. Perioli, F. Marmottini, I.D. Cunzolo, C. Rossi, Microporous and Mesoporous Mater. 96, 177–183 (2006)
J.L. Mi, C.H. Zhou, X. Bai, Chin. J. Antibiot. 32, 587–593 (2007). (in Chinese)
J.L. Mi, J. Wu, C.H. Zhou, West Chin. J. Pharm. Sci. 23, 84–86 (2008). (in Chinese)
Z.H. Chohan, S.H. Sumrra, M.H. Youssoufi, T.B. Hadda, Eur. J. Med. Chem. 45, 2739–2747 (2010)
S.K. Sridhar, S.N. Pandeya, J.P. Stables, A. Ramesh, Eur. J. Pharm. Sci. 16, 129–132 (2002)
L.X. Cheng, J.J. Tang, H. Luo, X.L. Jin, F. Dai, J. Yang, Y.P. Qian, X.Z. Li, B. Zhou, Bioorg. Med. Chem. Lett. 20, 2417–2420 (2010)
D.F. Xu, S.Z. Ma, G.Y. Du, Q.Z. He, D.Z. Sun, J. Rare Earths 26, 643–647 (2008)
Z.Y. Guo, R.G. Xing, S. Liu, Z.M. Zhong, X. Ji, L. Wang, P.C. Li, Carbohydr. Res. 342, 1329–1332 (2007)
G.Q. Hu, S.Q. Xie, G.J. Du, W.L. Huang, H.B. Zhang, S.T. Huang, Appl. Chem. 23, 273–277 (2006). (in Chinese)
C.O. Kappe, Curr. Opin. Chem. Biol. 6, 314–320 (2002)
B. Desai, D. Dallinger, C.O. Kappe, Tetrahedron 62, 4651–4664 (2006)
Y.S. Li, W.S. Yang, J. Membrane, Science 316, 3–17 (2008)
L.L. Liu, J. Yang, Z.G. Zhao, P.Y. Shi, X.L. Liu, J. Chem. Res. 34, 57–60 (2010)
X.Q. Wang, Z.G. Zhao, X.L. Liu, W.J. Li, J. Chem. Res. 34, 307–309 (2010)
J. Yang, Y.Y. Cheng, Z.C. Shi, Z.G. Zhao, J. Chem. Res. 34, 680–683 (2010)
G.H. Li, Z.C. Shi, X.R. Li, Z.G. Zhao, J. Chem. Res. 35, 278–281 (2011)
M.S. Karthikeyan, D.J. Poojary, B. Poojary, K.S. Bhat, B.S. Holla, N.S. Kumari, Bioorg. Med. Chem. 14, 7482–7489 (2006)
Z.L. Yuan, Q.H. Hu, Q. Wu, M.Q. Zhang, B.X. Zhu, Chin. J. Org. Chem. 29, 279–282 (2009). (in Chinese)
A.R.A.H. Farghaly, J. Chin. Chem. Soc. 51, 147–156 (2004)
X.Q. Wang, Z.G. Zhao, W.J. Li, X.L. Liu, T. Han, Chin. J. Org. Chem. 30, 764–767 (2010). (in Chinese)
Acknowledgments
This work was financially supported by the Science and Technology Bureau of Sichuan Province (no. 2012JY0028) and the Postgraduate Degree Construction Project of Southwest University for Nationalities of PR China (no. 2011XWD-S0703). We are thankful to the Southwest University for Nationalities management for their support and facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peng, Y., Zhao, Z., Liu, X. et al. Microwave-assisted synthesis and biological activity of new Schiff bases derived from dimers of 4-amino-3-[3-(1-benzyl)indole]-5-thiomethyl-1,2,4-triazole. Res Chem Intermed 39, 1897–1905 (2013). https://doi.org/10.1007/s11164-012-0723-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-012-0723-6